깨끗하고 그래핀으로 덮인 Cu(111) 표면에 흡착된 바나듐 원자의 전자적 특성은 초기 이론적인 방법을 사용하여 체계적으로 연구되었습니다. 이 작업에서는 바나듐 흡착의 두 가지 범위(1/9 ML 및 1 ML)를 고려합니다. 우리의 계산은 Cu 표면 아래에 머무는 V가 V/Cu(111)에 대해 앞서 언급한 두 가지 적용 범위에서 가장 안정적인 흡착 사이트인 것으로 밝혀졌습니다. 그러나 이러한 흡착은 바람직하지 않은 특성을 유발할 수 있습니다. 따라서 우리는 V와 Cu 표면 사이의 직접적인 상호 작용을 효과적으로 완화하기 위해 버퍼 층으로 그래 핀을 도입합니다. 계산은 원래 그래핀 층의 전자적 특성이 C 원자와 V 원자의 상호작용에 의해 크게 영향을 받는다는 것을 보여줍니다. 그래핀의 디랙 포인트는 두 적용 범위 모두에서 결과적으로 "파괴"됩니다. V/Gra/Cu(111) 시스템에서 그래핀 층과 기판 Cu 원자 사이의 상호 작용은 Gra/Cu(111) 시스템에서와 같이 약한 상태로 남아 있습니다. 또한, 1/9 ML의 상대적으로 낮은 적용 범위는 스핀 편극 시스템을 발생시키는 반면 비 스핀 편극 시스템은 1 ML의 적용 범위에서 관찰됩니다. 이 발견은 바나듐 기반 물질을 현실에 적용하는 새로운 방법을 제시합니다.
<섹션 데이터-제목="배경">
배경
불균일 촉매는 화학 및 에너지 산업의 많은 영역에서 중요한 역할을 합니다. 집중적인 연구는 지금까지 새로운 촉매를 이해, 개선 및 설계하는 데 중점을 두었습니다. 귀금속 기질에 대한 전이 금속 원자의 흡착은 해당 촉매 특성에 영향을 미칠 수 있으며, 이는 촉매 작용에서 가장 중요한 주제 중 하나입니다[1,2,3,4,5,6,7]. 특히, 금속 표면에 대한 하나의 단층 금속 흡착은 다양한 종류의 흡착 시스템 내에서 상당히 다른 화학적 및 촉매적 특성을 나타냅니다[5,6,7]. 일반적으로 물질의 촉매 특성은 원자 구조, 조성 및 페르미 준위에 가까운 전자 상태에 따라 달라집니다[8,9,10,11,12]. 기질은 금속 침착물의 촉매 특성에 직접 및/또는 간접적으로 영향을 미칠 것으로 예상됩니다. 우리 모두 알고 있듯이 Cu(111) 표면은 지난 수십 년 동안 가장 철저하게 조사된 단결정 금속 표면 중 하나입니다[13,14,15,16,17,18,19,20,21,22,23,24 ]. 특히, 지난 10년 동안 Cu(111) 표면은 CVD(Chemical Vapor Deposition)에 의한 고품질 및 대면적 그래핀의 성장을 위한 가장 중요한 기판으로 간주되어 왔다[22,23,24]. 그래핀의 새로운 전자적 특성은 이러한 기판에서 잘 보존될 수 있습니다. Cu(111) 표면에 후기 4d 전이 금속(Rh[25], Pd[26,27,28,29,30], Ir[31], Pt[29, 32, 33] 등)의 흡착 실험적으로나 이론적으로 폭넓게 연구되어 왔다. 그러나 Cu(111) 표면에 흡착된 초기 3차원 전이금속 원자에 대한 연구는 상대적으로 부족하다[34,35,36,37]. 여기에서는 이종 촉매, 분자 네트워크, 나노 재료 및 배터리 구축과 같은 여러 산업 분야에서 생화학적 관련성과 광범위한 응용으로 인해 초기 3d 전이 금속 원소인 바나듐에 중점을 둡니다[38]. 바나듐 기반의 다중음이온 물질은 상용 양극 물질인 LiCoO2를 대체할 후보로 제안되었습니다. 및 LiMn2 O4 유연한 원자가 상태로 인해 [39]. 따라서 바나듐 원자의 흡착 특성을 연구하면 실제 적용을 용이하게 할 수 있습니다. 연구된 시스템의 가능한 응용은 다음과 같이 예상할 수 있습니다. (1) 바나듐의 일반적인 산화 상태는 + 2, + 3, + 4 및 + 5일 수 있습니다. 따라서 나노재료 산업에서 강력하고 다재다능한 촉매로 사용될 수 있다[38]. (2) 금속 상태의 바나듐은 CO와 C의 불균형을 촉매하는 데 사용할 수 있습니다.2 [40]. (3) 전기 및 열 전도의 증가 가능성으로 인해 자유 전자 농도가 약한 표면에 흡착된 TM(즉, 바나듐) 원자를 분석하는 것도 흥미롭습니다[41]. 또한, 기록 매체, 자기 잉크 및 스핀트로닉 장치에 사용할 수 있는 2차원 표면 시스템의 자기 순서에 특별한 관심이 있습니다.
이 연구에서 우리는 밀도 기능 이론(DFT)을 기반으로 깨끗한 Cu(111) 표면과 그래핀으로 덮인 Cu(111) 표면에서 바나듐 원자의 흡착에 대한 체계적인 조사를 보고합니다. 위에서 언급한 두 시스템의 경우 전자 및 자기 특성에 대한 적용 범위의 영향을 평가하기 위해 바나듐 원자의 두 가지 대조적 적용 범위(즉, 1/9 ML 및 1 ML)가 고려됩니다. 깨끗한 Cu(111) 표면에 흡착된 V에 대한 가장 낮은 에너지 흡착 사이트는 V 범위에 관계없이 표면 위가 아니라 표면 아래에 있습니다. 그래핀으로 덮인 Cu(111) 표면에서 V의 흡착을 위해 흡착 사이트는 적용 범위에 따라 다릅니다. 1ML 적용 범위에 선호됩니다. 한편, V/Cu(111) 및 V/Gra/Cu(111) 시스템 모두에서 V 원자의 스핀 분극은 1/9 ML 적용 범위에 대해 에너지적으로 선호되는 반면 1 ML 적용 범위에서는 자성이 발견되지 않습니다. 또한 그래핀의 C 원자에 대한 순 자기 모멘트는 약 0.16μB입니다. /1/9 ML V/Gra/Cu(111) 시스템의 경우 탄소당, 이는 Gra/Cu(111) 시스템의 결과와 다릅니다. V/Cu(111) 및 V/graphene/Cu(111) 시스템의 상호 작용에 대한 깊은 이해를 얻기 위해 페르미 표면의 전자 상태를 자세히 분석합니다. 간단히 말해서, 우리의 연구는 V/Cu(111) 및 V/Gra/Cu(111) 시스템의 전자적 특성을 이해하는 데 도움이 될 수 있습니다.
방법
우리의 계산은 스핀-편광 밀도 기능 이론[43], 평면파 기반 및 프로젝터 증강파(PAW) 표현[44]에 기반한 비엔나 ab initio 시뮬레이션 패키지(VASP)[42]를 사용하여 수행되었습니다. ]. 일반화된 기울기 근사(GGA) 내의 Perdew-Burke-Ernzerhof(PBE) 교환 상관 에너지 함수[45]가 계산에 사용됩니다(B3LYP[46, 47] 및 HSE06[48] 하이브리드 함수를 사용한 일부 비교 연구도 다음과 같습니다. 필요할 때 제시). 그래핀과 Cu(111) 표면 사이의 반 데르 발스(vdWs) 상호 작용을 정확하게 설명하기 위해 vdWs 보정 기능이 있는 PBE(DFT-D2)[49]가 채택되었습니다. 컷오프 평면파 운동 에너지는 500eV로 설정됩니다. Cu(111) 표면은 약 20 Å의 진공 간격과 함께 7개의 Cu 층으로 구성된 슬래브 모델을 사용하여 모델링되었습니다. Cu(111) 및 Gra/Cu(111) 표면의 서로 다른 V 적용 범위는 서로 다른 슈퍼셀을 사용하여 모델링되었습니다. 1/9 ML 및 1 ML의 V 적용 범위에 대해 각각 (3 × 3) 및 (1 × 1) 표면 단위 셀을 사용했습니다. 24 × 24 × 1 k-가 있는 Monkhorst-Pack 방식 [50] 메쉬는 (1 × 1) 표면 단위 셀에 대한 Brillouin 영역 통합을 샘플링하는 데 사용되었으며 8 × 8 × 1 k- 메쉬는 (3 × 3) 표면 단위 셀에 사용되었습니다. 최적화하는 동안 슬래브의 가장 낮은 세 개의 Cu 레이어는 동결된 반면 시스템의 나머지 원자는 각 원자에 가해지는 힘이 0.01eV/Å 미만이 될 때까지 완전히 이완되었습니다. 바나듐 원자는 슬래브의 한 면에 흡착되었습니다. 쌍극자 보정[51]은 우리의 계산을 기반으로 발견된 무시할 수 있는 에너지 보정으로 인해 이 연구에서 고려되지 않습니다.
결과 및 토론
깨끗한 Cu(111) 표면의 바나듐 원자 흡착
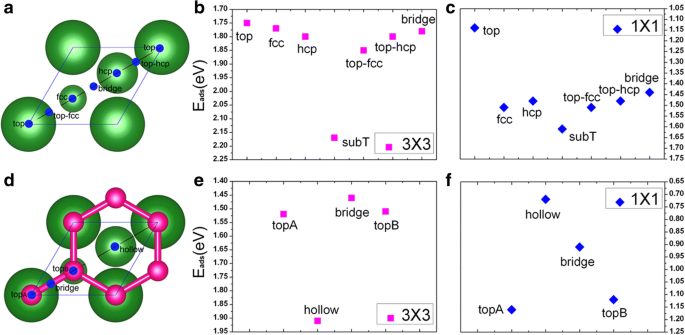
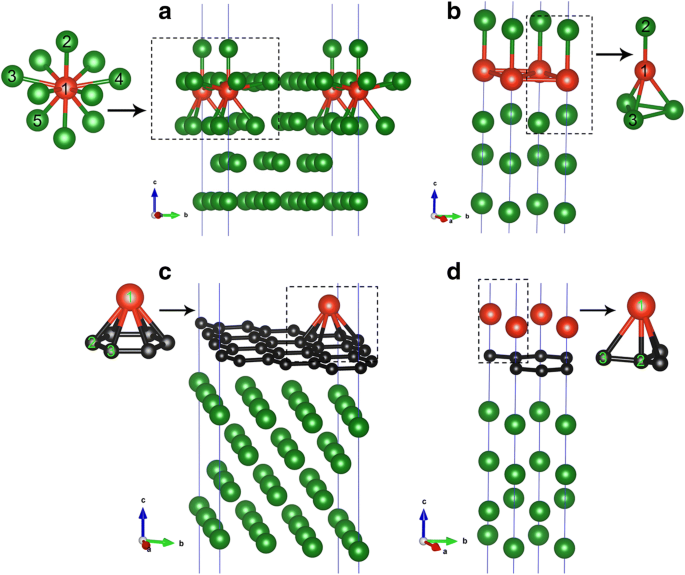
이 섹션에서는 두 가지 적용 범위(즉, 1/9 ML 및 1 ML)에서 깨끗한 Cu(111) 표면에 직접 V 흡착에 대한 결과를 제시합니다. Cu(111) 표면에서 V 원자의 유리한 흡착 사이트를 찾기 위해 각 커버리지에 대해 가능한 7개의 가능한 흡착 사이트, 즉 top, fcc, hcp, subT, top-fcc, top-hcp 및 브리지 사이트를 고려합니다. , 도 1a에 도시된 바와 같이. 특히, subT는 V 원자가 표면층 Cu 원자와 위치를 교환하는 Cu 표면 아래의 사이트라는 점에 유의해야 합니다(그리고 Cu 원자를 바나듐 원자 바로 위의 사이트로 이동). 그림 2a, b 참조. 두 흡착 범위에 대해 V/Cu(111) 시스템에 대해 7개 흡착 사이트 모두에 대한 흡착 에너지가 계산됩니다. 얻어진 결과는 그림 1b, c에 나와 있습니다. 여기서, 바나듐 원자당 흡착 에너지(Ead )는 다음 공식으로 계산됩니다.
$$ {\mathrm{E}}_{\mathrm{ad}}=\left[\left({NE}_V+{E}_{Cu(111)}\right)-{E}_{V/ Cu (111)}\오른쪽]/N $$
여기서 EV 고립된 바나듐 원자의 에너지, E쿠 (111) 관련된 깨끗한 Cu(111) 표면의 총 에너지, EV /쿠 (111) V/Cu(111) 시스템의 총 에너지, N 관련된 V 원자의 수입니다. 그림 1b, c에서 subT 사이트는 위에서 언급한 커버리지에 대해 Cu(111) 표면의 V 흡착에 대해 에너지적으로 선호됨을 알 수 있습니다. 이를 바탕으로 다음 논의에서는 subT 사이트만을 고려할 것이다. 계산된 흡착 에너지, V 원자와 인접한 Cu 원자 사이의 결합 길이, V/Cu(111)에 대한 V 원자의 원자 자기 모멘트는 표 1에 나열되어 있습니다. 표 1에서 볼 수 있듯이 흡착 에너지는 E광고 1/9 ML 및 1 ML 적용 범위에 대해 각각 V 원자당 2.17 및 1.61 eV이며, 이는 V 원자와 Cu(111) 표면의 상호 작용이 매우 강함을 나타냅니다. 더욱이, 흡착 에너지는 V 범위가 증가함에 따라 감소하는데, 이는 V-V 상호작용이 더 강해지고 V 층과 Cu 표면 사이의 상호작용이 약해진다는 것을 의미한다. V 원자와 인접한 Cu 원자 사이의 가장 짧은 결합 길이는 1/9 ML 및 1 ML 적용 범위에 대해 각각 2.27 및 2.37 Å입니다. 이것은 V 원자와 Cu 기판 사이의 상호작용이 1/9 ML에 대해 상대적으로 강함을 의미하며, 이는 흡착 에너지로부터 계산된 결과와 일치합니다. V 원자의 강자성(FM) 차수도 계산에 고려되며 FM 차수의 스핀 분극 에너지는 ΔE로 계산됩니다. =(E아니요 _잡기 − EFM )/N (E 포함 아니요 _잡기 비자기 상태의 에너지). 바나듐 원자의 스핀 분극 에너지는 1/9 ML 적용 범위에 대해 110 meV인 반면(표 1 참조) 1 ML 적용 범위에는 자성이 없습니다. V의 원자 자기 모멘트는 1.34μB입니다. 값과 매우 다른 바나듐의 1/9 ML 적용 범위(3 μB ) 기상 V 원자. 이 점에 대해서는 나중에 논의하겠습니다.
<그림>
아 깨끗한 Cu(111) (1 × 1) 표면의 흡착 사이트:가장 큰 볼은 표면 Cu 원자를 나타내고 작은 볼은 하위층 Cu 원자를 나타냅니다. ㄴ , ㄷ 각각 1/9 ML 및 1 ML 적용 범위에서 Cu(111)에 대한 V 원자의 서로 다른 흡착 위치의 흡착 에너지; d 그래핀으로 덮인 Cu(111) (1 × 1) 표면, 빨간색 볼은 그래핀의 C 원자를 나타냅니다. , f 각각 1/9 ML 및 1 ML 범위에서 그래핀으로 덮인 Cu(111)에 대한 V 원자의 흡착 에너지
<그림>
a에 대한 V/Cu(111) 시스템의 기하학 1/9ML 및 b 1ML 적용 범위. c에 대한 V/Gra/Cu(111) 시스템의 기하학 1/9 ML 및 d V 원자의 1ML 적용 범위. 빨간색, 검은색 및 녹색 볼은 각각 V, C 및 Cu 원자를 나타냅니다.
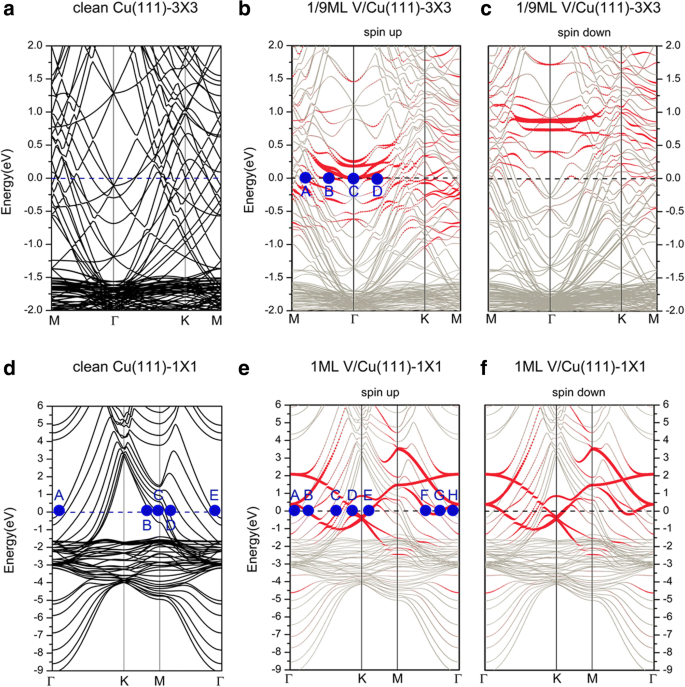
다음으로 V/Cu(111) 시스템의 전자 구조에 대해 논의합니다. Cu(111)(3 × 3) 및 Cu(111)(1 × 1) 표면(즉, 1/9 및 1 ML)에 대한 V 흡착의 밴드 구조는 그림 3에 나와 있으며, 상응하는 깨끗한 Cu(111)(3 × 3) 및 Cu(111)(1 × 1) 표면도 비교를 위해 표시됩니다. 그림 3a, d 모두 깨끗한 Cu(111)의 전자 구조에 대해 논의하는 데 사용할 수 있습니다. 우리는 여기에서 선택합니다. 그림 3d. 전자 및 d Cu의 전자는 깨끗한 Cu(111) 표면을 위한 시스템의 전도도에 기여합니다. 더 자세하게, 우리는 그림 3d의 페르미 표면에 대표 점(A, B, C, D, E)을 표시했습니다. 포인트 A와 E는 주로 dyz 표면 Cu 원자의 전자. 포인트 B와 C는 d의 기여도를 보여줍니다. xy 그리고 dx2− y2
각각 Cu 원자의 전자. 포인트 D는 의 혼합을 설명합니다. d가 있는 전자 z2
그리고 dx2− y2
인접한 Cu 원자 사이의 전자. V가 Cu(111)에 흡착되면 얻은 밴드 구조는 V 커버리지가 변화함에 따라 다른 방식으로 변화합니다. 1/9 ML 적용 범위(그림 3b, c 참조)의 경우 스핀-업 및 스핀-다운 채널의 밴드 구조가 다르며 이는 스핀 편광 기능을 나타냅니다. 그림 3에서 빨간색 점은 V 원자의 기여를 나타내고 은회색 점은 배경 Cu의 기여를 나타냅니다. 스핀업 채널(즉, 다수 스핀)에서 d V 원자의 전자와 기질 Cu 원자는 페르미 표면의 전자 상태에 크게 기여합니다. d의 교잡 표면 Cu 원자와 V 원자의 전자가 명확하게 보입니다. 명확하게 설명하기 위해 그림 3b의 페르미 표면에 몇 가지 대표적인 점(A, B, C, D)도 표시했습니다. 거기에서 점 A는 d의 혼합을 보여줍니다. z2d를 갖는 V 원자의 전자 yz , dz2
표면 Cu 원자의 전자. 모든 B, C 및 D 포인트는 d의 기여도를 나타냅니다. V 원자의 전자. 예를 들어, 포인트 B는 dx2-y2
V 원자의 전자. 스핀다운 채널(즉, 소수 스핀)의 경우 밴드 구조는 V 원자에서 기여한 전자 상태가 모두 페르미 준위(비점유)보다 훨씬 높다는 것을 보여줍니다. 페르미 표면에 대한 기여는 주로 s , d Cu 원자의 전자, V 원자의 전자로부터 아주 작은 기여. 이 두 스핀 채널 사이의 존재하는 차이는 V 원자에 대한 자기 모멘트를 나타냅니다(1.34μB ). 1 ML 적용 범위(그림 3e, f 참조)의 경우 1/9 ML의 상황과 달리 흡착된 시스템은 스핀 극성이 없습니다. 편의상 그림 3e의 페르미 표면에 대표점(A, B, C, D, E, F, G, H)도 표시했습니다. 점 A와 H는 모두 d의 기여도에 해당합니다. yz Cu 원자의 최하층 전자. 지점 B, D, E, F 및 G의 전자 상태는 d에 의해 제공됩니다. V adatom의 전자. 예를 들어, dx2− y2
그리고 dxz 의 V 원자는 각각 B와 D의 전자 상태에 기여합니다. 페르미 표면의 B와 D 사이의 점에 대해 우리는 d의 복잡한 혼합을 발견했습니다. s, d가 있는 V 원자의 전자 V 원자 주변의 Cu 원자의 전자. 예를 들어, C 지점의 경우 전자 구조는 s, dyz, 그리고 dz2
표면층 Cu의 전자와 dz를 갖는 최상층 Cu 원자
2
V 원자의 전자.
<그림>
a의 브릴루앙 구역(BZ)에 그려진 깨끗한 Cu(111) 표면의 밴드 구조 3 × 3 단위 셀 및 d 1 × 1 단위 셀. b에 대한 1/9 ML 적용 범위의 Cu(111) 표면에 대한 V 흡착의 밴드 구조 스핀업 및 c 스핀 다운. e에 대한 1ML 범위의 Cu(111) 표면에 대한 V 흡착의 밴드 구조 회전 및 f 스핀다운
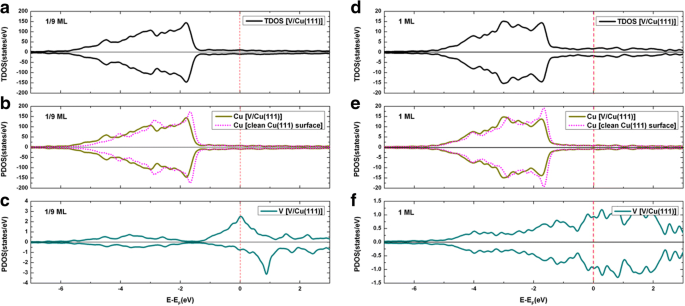
깨끗한 Cu(111) 표면에 대한 V 흡착의 전체 상태 밀도(TDOS)는 예상 밀도(PDOS)와 함께 1/9 ML 및 1 ML 적용 범위 모두에 대해 그림 4에 나와 있습니다. 분명히, V 원자의 명백한 스핀 분극은 1/9 ML에서 발견되는 반면(그림 4c 참조) V 원자의 스핀 분극은 1 ML에서 발견되지 않습니다(그림 4f 참조). 또한, 1/9 ML 및 1 ML 적용 범위 모두에서 Cu 원자에 대해 스핀 분극이 관찰되지 않습니다(그림 4b, e 참조). V 흡착 전후에 Cu의 PDOS를 통합함으로써(즉, Cu의 전자 수로 이어짐) Cu 원자의 전하가 약간 증가한다는 것을 발견했으며, 이는 V 원자에서 Cu 기판으로의 전하 이동을 나타냅니다. V/Cu(111). 즉, V 흡착은 Cu에 n형 도핑을 유도합니다. Cu(111) 표면의 V 흡착을 더 잘 이해하기 위해 그림 5a, b에 각각 1/9 ML 및 1 ML 적용 범위에서 변형 전하 밀도의 등고선 그림을 그립니다. 변형 전하 밀도는 \( \Delta \rho \left(\overrightarrow{r}\right)={\rho}_{\left[V/ Cu(111)\right]}\left(\overrightarrow{ r}\right)-\sum \limits_{\mu =1}^N{\rho}^{atom}\left(\overrightarrow{r}-\overrightarrow{R_{\mu }}\right) \). 그림 5에서 볼 수 있듯이 V 원자와 인접한 Cu 원자 사이의 공유 및 이온 결합은 1/9 ML 및 1 ML 적용 범위 모두에서 명확하게 볼 수 있습니다. 특히 공유 결합은 1/9 ML 적용 범위(1 ML과 비교할 때)에서 상대적으로 더 강한 반면 이온 결합은 1 ML 적용 범위에서 상대적으로 더 강합니다.
<그림>
아 1/9 ML 적용 범위에서 V/Cu(111) 시스템의 TDOS; ㄴ 1/9 ML에서 기질의 전체 Cu 원자의 PDOS; ㄷ 1/9 ML에서 V adatom용 PDOS; d 1ML에서 V/Cu(111) 시스템의 TDOS; 이 1 ML에서 기질의 전체 Cu 원자에 대한 PDOS; 및 f 1ML에서 V adatoms용 PDOS. 특히, 깨끗한 Cu(111) 표면의 DOS는 그래프 b에도 주입됩니다. 및 e 비교를 위해
<그림>
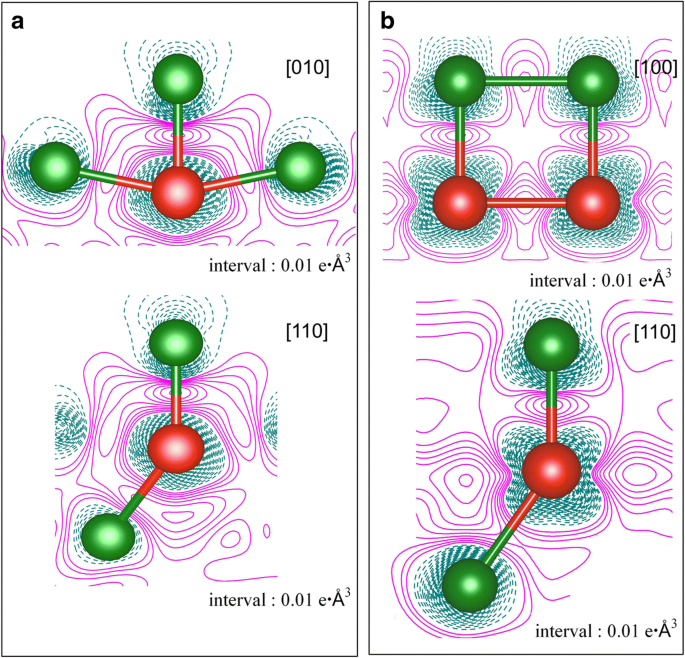
두 가지 적용 범위, 즉 a에서 Cu(111) 표면의 V 원자 흡착에 대한 변형 전하 밀도 1/9ML 및 b 1ML. 전자 축적 및 고갈은 각각 자홍색 실선과 진한 청록색 대시선으로 표시됩니다. 녹색 및 빨간색 볼은 각각 Cu 및 V 원자를 나타냅니다.
Cu(111) 표면의 그래핀 층
subT 사이트는 위의 논의에서 흡착 에너지로부터 깨끗한 Cu(111)의 V 원자에 대한 가장 안정적인 흡착 사이트인 것으로 밝혀졌습니다. 이러한 흡착 사이트는 일부 관심 대상이지만; 그러나 Cu 표면층 아래에 머무르는 V 원자는 표면에서 촉매로서의 적용을 제한하는 바람직하지 않은 특성을 초래할 수 있습니다. 따라서 V 원자와 Cu(111) 표면 사이의 직접적인 상호작용을 완화하기 위해 버퍼층을 도입하려고 합니다. 그래핀은 본 연구에서 가장 먼저 고려된 V/Cu(111) 시스템과 가장 근접한 격자 일치로 인해 완벽한 선택입니다.
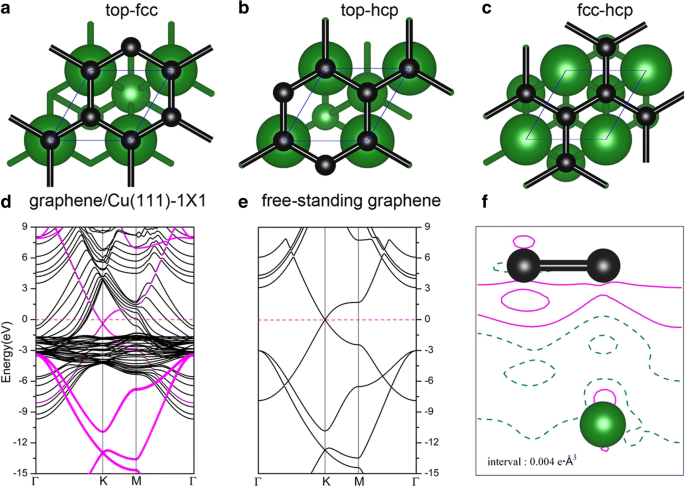
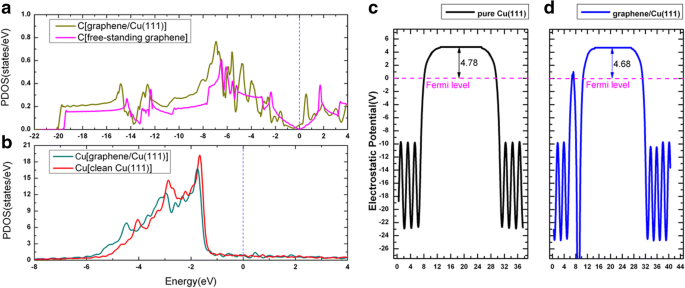
금속 표면에 대한 그래핀의 흡착은 여러 이전 출판물에서 집중적으로 연구되었습니다[52,53,54]. 그래핀이 Cu(111)에 흡착될 때 그래핀/Cu(111) 시스템(이하 Gra/Cu(111)으로 가명)의 세 가지 가능한 기하학적 구조가 고려되었습니다. 즉, 그래핀은 top-fcc, top-hcp에 있었습니다. 및 fcc-hcp 흡착 부위; 그림 6a-c 참조. 우리의 결과를 기반으로, top-fcc 기하학(그림 6a)은 탄소 원자당 47 meV의 흡착 에너지와 함께 에너지적으로 가장 안정적인 구조로 표시되며 그래핀과 Cu(111) 표면 사이의 평형 거리는 3.14입니다. Å, 이는 이전 연구[52,53,54]와 상당히 일치합니다. 이러한 낮은 흡착 에너지(47 meV/C)와 큰 층간 거리는 그래핀과 Cu(111) 사이의 결합이 상대적으로 약하다는 것을 의미한다. 그림 6d는 top-fcc 구성을 가진 그래핀/Cu(111) 시스템의 밴드 구조를 보여줍니다(그림 6a). 특히 그래핀에서 제공하는 전자 상태는 그림에서 자홍색 원으로 스케치됩니다. 독립형 그래핀 시트의 밴드 구조도 비교를 위해 그림 6e에 나와 있습니다. 이 그림에서 알 수 있듯이, 그래핀의 밴드 구조는 독립 시트와 Cu(111) 표면의 시트 사이에서 매우 유사합니다. K의 Dirac 지점(선형 밴드 교차 포함)은 그림 6d에서 보존되지만 그래핀이 Cu(111) 표면에 흡착될 때 약간의 하향 이동이 있습니다. 교차점의 하향 이동은 Cu(111) 기판에서 그래핀 층으로의 전하 이동을 나타내며, 이는 Al, Ag 및 Cu가 그래핀에 의해 n형 도핑된다는 이전 결과에 따른 것입니다[52,53,54 ]. 변형 전하 밀도, 즉 Gra/Cu(111)의 총 전하 밀도와 독립 그래핀과 깨끗한 Cu(111) 표면의 전하 밀도 합 간의 전하 차이, 즉 \( \Delta \rho \ left(\overrightarrow{r}\right)={\rho}_{Gra/Cu(111)}\left(\overrightarrow{r}\right)-{\rho}_{Gra}\left(\overrightarrow{ r}\right)-{\rho}_{Cu(111)}\left(\overrightarrow{r}\right) \)는 그림 6f와 같다. 그림 6f에서 볼 수 있듯이 C 원자 주변의 실선에 따라 Cu(111) 기판에서 그래핀 층으로의 전하 이동도 관찰할 수 있습니다. 한편, 우리는 Gra/Cu(111) 시스템에서 C 및 Cu 원자에 대한 PDOS(projected density of state)를 그림 1에서 Cu(111) 표면(그래핀 흡착 유무)의 일함수 변화와 함께 플로팅합니다. 7. PDOS 적분을 통해 C 원자의 전자는 약간 증가하고 Cu 원자의 전자는 약간 감소하여 전하 이동 현상을 확인했습니다. 더욱이, 우리는 그래핀 층의 흡착 후에 Cu(111)의 계산된 일함수가 4.78에서 4.68eV로 변한다는 것을 발견했다. 이 모든 것은 전하 이동이 Cu 기판에서 그래핀 층으로 이동한다는 것을 확인시켜줍니다.
<그림>
Cu(111) 표면의 그래핀 시트 형상:a 상단 fcc, b top-hcp 및 c fcc-hcp 사이트. de와 비교한 top-fcc 기하학의 그래핀/Cu(111) 시스템의 밴드 구조 자립형 그래핀. 마젠타색 점은 그래핀이 기여하는 전자 상태를 나타냅니다. 에 그래핀/Cu(111)의 총 전하 밀도와 top-fcc 기하학에 대한 독립 그래핀 및 깨끗한 Cu(111) 표면의 전하 밀도 합계 간의 전하 차이. 등고선의 간격은 0.004e Å
−3
입니다.
<그림>
a에 대한 그래핀/Cu(111) 상태의 예상 밀도 C 원자 및 b 구리 원자. c, d 깨끗한 Cu(111) 표면과 그래핀/Cu(111) 계면의 일함수
그래핀으로 덮인 Cu(111) 표면의 바나듐 흡착
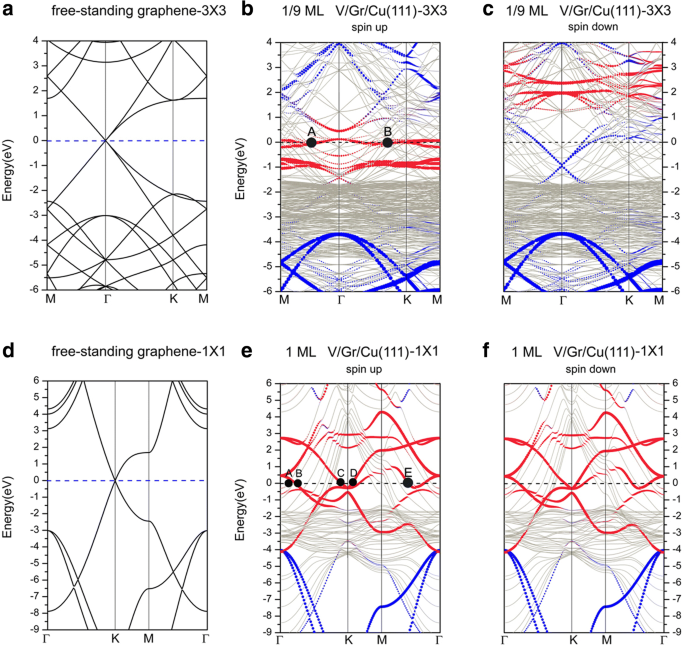
이 섹션에서는 그래핀으로 덮인 Cu(111) 표면에서 바나듐 흡착의 원자, 전자 및 자기 특성을 평가하려고 합니다. 그림 1d와 같이 topA, Bridge, topB 및 중공 사이트로 표시된 4개의 가능한 흡착 사이트가 고려됩니다. 1/9 ML 및 1 ML에서 Gra/Cu(111)에 대한 V 원자의 흡착 에너지는 각각 그림 1e, f에 나와 있습니다. 그래핀으로 덮인 Cu(111) 표면의 V 흡착에 대해 에너지적으로 선호되는 사이트는 적용 범위에 따라 다릅니다. 보다 구체적으로, V adatom은 1/9 ML에 대해 최대로 배위된 중공 부위(그림 1d 참조)를 선호하는 반면, 낮은 배위된 상단 부위(즉, topA 부위, 그림 1d 참조)는 1ML의 높은 적용 범위에 대해 선호됩니다. . V/Gra/Cu(111) 시스템에 대한 흡착 에너지, V 원자와 인접한 C 원자 사이의 결합 길이, 바나듐과 탄소의 원자 자기 모멘트가 표 1에 나열되어 있습니다. Gra/Cu(111) 표면, Ead , 1/9 ML 및 1 ML에 대해 각각 V 원자당 1.91 및 1.16 eV이며, 이는 Cu(111) 표면의 것과 비교할 때 어느 정도 감소합니다. 분명히 그래핀 버퍼층의 도입은 우리가 예상한 대로 V 원자와 Cu(111) 표면 사이의 상호 작용을 약화시킬 수 있습니다. 우리는 다른 범위에서 V/Gra/Cu(111)에서 V의 강자성 차수의 스핀 분극 에너지를 더 조사합니다. 스핀 분극 에너지는 1/9ML에 대해 390meV인 반면 1ML에는 스핀 분극이 없습니다. 스핀 분극 에너지는 V/Cu(111) 시스템에서와 비교할 때 V/Gra/Cu(111) 시스템에서 상당히 더 높습니다(110 meV와 비교하여 390 meV, 표 1 참조). 1/9 ML에서 V/Gra/Cu(111) 시스템에서 V 원자의 자기 모멘트는 2.93 μB입니다. 3μB에 가깝습니다. /atom(기체상 V 원자의 값)은 V 원자가 잘 분리되어 있고 V 원자와 그래핀 층 사이에 전달되는 전하가 거의 없음을 의미합니다. C 원자에 대한 작은 자기 모멘트(0.16μB /atom)도 있습니다.
다음으로, 그래핀으로 덮인 Cu(111) 표면에서 V 흡착의 밴드 구조에 대해 논의합니다. 그림 8은 Gra/Cu(111) 표면의 V 흡착 밴드 구조와 두 개의 다른 단위 셀에 그려진 독립형 그래핀의 밴드 구조를 보여줍니다. 여기서 파란색과 빨간색 원은 각각 그래핀과 V 아톰의 기여도를 나타냅니다. 첫째, 페르미 준위를 가로지르는 많은 Cu 밴드가 있다는 점에 유의해야 합니다. 이는 시스템의 Cu 원자가 시스템의 전도도에 크게 기여함을 나타냅니다. 1/9 ML 적용 범위의 경우 밴드 구조(그림 8b, c 참조)는 시스템의 전자 구조가 스핀 분극화되어 있음을 보여줍니다. 이전 섹션에서 언급했듯이 그래핀과 Cu(111) 표면 사이의 상호 작용은 Gra/Cu(111)에 대해 매우 약합니다. 따라서 그래핀이 기여하는 밴드는 전체 밴드 구조에서 쉽게 인식됩니다. 그러나 그래핀으로 덮인 Cu(111) 표면(즉, V/Gra/Cu(111) 시스템)에 V 흡착 후 그래핀의 Dirac 점이 완전히 "파괴"되었음을 알 수 있습니다(그림 8b 참조). 스핀업 채널의 밴드 구조와 그래핀의 "선형 교차점"은 스핀다운 채널에서 여전히 구별할 수 있습니다. 그럼에도 불구하고, 스핀다운 구성요소에서 "선형 교차점"의 매우 많은 하향 이동은 그래핀 층으로의 전하 이동이 상대적으로 많다는 것을 나타냅니다. C 층과 Cu 층 사이의 상호 작용이 약하기 때문에 그래핀 층으로 전달된 전하는 V 원자 층에서 유래한다는 점에 유의해야 합니다(그림 10a 참조). 1/9 ML의 스핀업 채널의 경우 d 기판의 Cu 원자의 전자는 페르미 표면에 기여하며 d도 많이 있습니다. V adatom의 전자가 시스템의 전도도에 기여합니다. 한편, pd가 모두 있는 C 원자의 전자 V 원자 및 표면 Cu 원자의 전자가 보입니다(그러나 현저하지는 않음). 마찬가지로 그림 8b의 페르미 표면에 두 개의 대표적인 점(A, B)을 표시했습니다. 포인트 A는 d의 기여도를 나타냅니다. xy , dx2− y2
V adatom의 전자, 점 B는 p의 혼성화에 대한 기여를 보여줍니다. zd가 모두 있는 C 원자의 전자 z2
V 원자의 전자와 Cu 원자의 최상층. 분명히 표면 V층은 중요한 전도층입니다. 대조적으로, 1/9 ML의 스핀다운 채널에서 페르미 수준의 전자 상태는 주로 d Cu 원자 및 p의 전자 z C 원자의 전자; d의 기여 V adatom의 전자는 무시할 수 있습니다. 1 ML 적용 범위의 경우 그림 8e, f에 표시된 밴드 구조는 시스템이 스핀 편극되지 않음을 의미합니다. 따라서 그래핀 층의 Dirac 지점도 "파괴"되기 때문에 V 원자와 그래핀 버퍼 층 사이의 상호 작용이 강해야 합니다. 시스템의 계산된 전자 상태에서 페르미 준위에 기여한 전자는 주로 s , d Cu 및 d의 전자 V 원자의 전자와 p -C 원자의 전자. 더 자세하게, 페르미 표면에 위치한 k-포인트 A, B, C, D, E는 그림 8e에 표시되어 있다. 점 A는 d의 혼성화를 나타냅니다. yz 그리고 dz2
첫 번째 층(최상위) Cu 원자. 점 B, C 및 D는 d의 전자 상태입니다. - V adatom의 전자. 보다 구체적으로, 점 B는 d의 전자 상태를 나타냅니다. xy , dx2− y2
V adatom의 전자. 또한, 포인트 E는 s의 강한 혼성화를 보여줍니다. , dz2p가 있는 V 원자의 전자 zp의 상대적으로 약한 혼합과 함께 C 원자의 전자 zd를 갖는 C 원자의 전자 z2
Cu 원자의 최상층의 전자.
<그림>
Band structures of a free-standing graphene, plotted in the Brillouin zones of a a (3 × 3) unit cell and d a (1 × 1) unit cell. Band structures of V adsorptions on the graphene-covered Cu(111) surfaces for b spin up of 1/9 ML, c spin down of 1/9 ML, e spin up of 1 ML, and f spin down of 1 ML
We now present the density of states for the V/Gra/Cu(111) system. The total density of states of V adsorption on the graphene-covered Cu(111) surface, together with the projected densities of states, are demonstrated in Fig. 9 for both the 1/9 ML and 1 ML coverages. At 1/9 ML, the spin polarizations of V adatoms and C atoms in the graphene layer are clearly seen (see Fig. 9c, d), while no spin polarization is found for Cu atom (see Fig. 9b). At 1 ML coverage, no spin polarization has been found for all atoms (see Fig. 9f-h). At 1/9 ML coverage, the DOS of the spin-up channel at the Fermi level is mainly contributed from the Cu atoms (totally 11.9 states/eV∙u.c.) and V atoms (totally 5.8 states/eV∙u.c.), with only minor contributions from the graphene layer (totally 0.4 states/eV∙u.c.). Meanwhile, the DOS of spin-down channel at the Fermi level is mainly contributed from the Cu atoms (totally 11.9 states/eV∙u.c.) and graphene layer (totally 1.1 states/eV∙u.c.), with only minor contributions from the V atoms (totally 0.1 states/eV∙u.c.). For the 1 ML coverage, both the DOS of spin-up and spin-down channels at the Fermi level are mainly contributed from the Cu atoms and V atoms (i.e., 1.1 and 0.7 states/eV∙u.c. for each spin component, respectively), with negligible contribution from the graphene layer (0.04 states/eV∙u.c). By integrating the PDOSs for each atom before and after the V adsorptions (leading to number of electrons), the charge transfer can be determined for different atoms. To be specific, the total valence electrons of the Cu atoms are reduced slightly for both the 1/9 ML and 1 ML coverages when compared with those of a clean Cu(111) surface, while the total valence electrons of C atoms are slightly increased when compared with that of a free-standing graphene. This implies that small amount of charges are transferred from Cu substrate to graphene layer for V/graphene/Cu(111) systems regardless of the V coverages. The total valence electrons of Cu atoms in V/Gra/Cu(111) systems are almost equal to those in the graphene/Cu(111) systems, which indicates that the Cu substrate has not been affected by V adsorption. The physical pictures given by the analysis of DOSs here are all in consistent with the analysis of the band structures. Finally, we show in Fig. 10 the contour plots of the deformation charge densities for the 1/9 ML and 1 ML coverages, respectively. The deformation charge density is defined as \( \Delta \rho \left(\overrightarrow{r}\right)={\rho}_{\left[V/ Gra/ Cu(111)\right]}\left(\overrightarrow{r}\right)-\sum \limits_{\mu =1}^N{\rho}^{atom}\left(\overrightarrow{r}-\overrightarrow{R_{\mu }}\right) \). As shown in Fig. 10, the interactions between the graphene layers and the substrate Cu atoms are both relatively weak for 1/9 ML and 1 ML coverages, which are in consistent with the above discussions. From Fig. 10a, for the 1/9 ML, the bonding between V adatoms and its adjacent C atoms is mainly ionic, and the covalent bonding is not obvious. In contrast, for the 1 ML coverage, both ionic and covalent bonding between V adatom and its adjacent C atoms are clearly visible (see Fig. 10b). Besides, the covalent bonding between neighboring V adatoms is also very significant at 1 ML coverage. Due to the existence of graphene buffer layer, V adatoms cannot interact directly with the Cu atoms.
아 TDOS of V adsorption on graphene-covered Cu(111) surface at 1/9 ML coverage; PDOS for b Cu atoms, c C atoms, and d V adatoms at 1/9 ML. Likewise, e TDOS of V adsorption on Gra/Cu(111) surface at 1 ML coverage; PDOS for f Cu atoms, g C atoms, and h V adatoms at 1 ML. For comparison, PDOS of Cu atoms of a clean Cu(111) and graphene/Cu(111) are implanted in b and f , while PDOS of C atoms of a free-standing graphene are also implanted in c and g
Deformation charge densities for the adsorption of V atoms on the graphene-covered Cu(111) surface at two coverages, i.e., a 1/9 ML and b 1 ML. Electron accumulation and depletion are represented by magenta solid lines and green dashed lines, respectively. The green, black, and red balls represent Cu, C, and V atoms, respectively
We have also calculated the phonon spectra for both the V/Cu(111) and V/Gra/Cu(111) systems. From the calculated phonon spectra, we find that there is no “imaginary frequency” for both the two types of systems, indicating that the systems studied are dynamically stable and would be seen in the laboratory. Since the main purpose of our work is not the thermodynamic stability, therefore the figures of the phonon dispersions are not shown in this text. Second, we have noticed that the different DFT functionals we adopted may lead to the different results. Hence, we have calculated the 1 ML V/Gra/Cu(111) system (as a representative) within the DFT framework under the B3LYP, HSE06 hybrid functionals, as well as the PBE functional. The results suggest that the adsorption site with largest adsorption energy is the topA site, calculated from all the PBE, HSE06, and B3LYP methods. However, relative adsorption energies at different adsorption sites from the B3LYP and PBE and HSE06 methods differ significantly (results from PBE and HSE06 methods are almost the same, since this is a metallic system). On the other hand, the geometrical parameters obtained from the three functionals show good consistency. Although the detailed charge density contours are somewhat different between PBE and B3LYP method, the main bonding characteristics are the same from both the two methods. In summary, the main point is that the adsorption energies obtained from B3LYP functional are significantly larger than those from the PBE and HSE06 functionals. To explain this point, Paier et al. argued that B3LYP functional lacked of a proper description of the “free-electron-like” systems with a significant itinerant character (e.g., metals and small gap semiconductors). They have concluded that the overestimation of the total energy of the atoms can be induced by the significantly overestimation on the exchange and correlation energies of B3LYP functional. In this respect, PBE functional often shows much more reliable results [55].
결론
To summarize, using first-principles calculations, we have systematically investigated the electronic and geometric properties of the adsorption of V atoms on both the clean Cu(111) surface and the graphene-covered Cu(111) surface. Firstly, for the V/Cu(111) system, an adsorption site underneath the Cu surface layer is found as the preferable adsorption site for V atom regardless of the coverages. The hybridization of V’s d states with Cu’s d states rules the electronic properties of V/Cu(111) systems. Ferromagnetic order of V adatoms is energetically favored for 1/9 ML coverage (1.34 μB /atom), while no magnetism of V adatoms is observed for 1 ML coverage. Due to the strong interaction between V adatom and its adjacent substrate’s Cu atoms, the magnetic moment of V is significantly reduced. Secondly, the graphene/Cu(111) systems are investigated and the results agree well with the previous literatures. Thirdly, adsorptions of V on the graphene-covered Cu(111) at two coverages (i.e., 1/9 ML and 1 ML) show different preference of adsorption sites. The hollow site with maximum coordination is energetically favored for the adsorption of 1/9 ML, while the top site with low coordination is preferred for 1 ML adsorption. In V/Gra/Cu(111) systems, the interactions of C atoms with the V adatoms destroy the electronic properties of both the original graphene layer and the adsorbed atoms, represented by the strong hybridization of C’s pz -states with V adatoms’ dz2
-states. A net magnetic moment for C atoms of graphene also appeared (0.16 μB/per carbon). In short, our study paves the way to a deep understanding of the adsorption properties of vanadium atoms on the clean Cu(111) and graphene-covered Cu(111) substrates. Simultaneously, this study also provides a reference for the possible applications of the V/Cu(111) and V/Gra/Cu(111) systems in the catalyst in nanomaterials industry, spintronic devices, and others.
약어
Cu(111):
(111) surface of copper
FM:
Ferromagnetic
GGA:
Generalized gradient approximation
ML:
Monolayer
PAW:
Projector augmented wave
PBE:
Perdew-Burke-Ernzerhof
V/Cu(111):
Vanadium atoms adsorbed on Cu(111) surface
V/Gra/Cu(111):
Vanadium atoms adsorbed on graphene-covered Cu(111) surface