

나노물질

산업 제조
두개내 동맥류(IA)에서 일부 긴 비암호화 RNA(lncRNA)의 역할은 많은 연구에서 조사되었습니다. 이 연구의 목적은 혈관 내피 손상 유발 IA에서 lncRNA 전이 관련 폐 선암종 전사체 1(MALAT1)/microRNA-143(miR-143)/혈관 내피 성장 인자-A(VEGFA) 신호 축의 메커니즘을 밝히는 것입니다. . IA 조직 및 정상 동맥 조직에서 MALAT1, miR-143 및 VEGFA 발현이 검출되었습니다. 조직에서 기질 금속단백분해효소 9(MMP-9), 혈청 및 조직에서 vWF(von Willebrand factor), 혈청에서 엔도텔린-1(ET-1)이 검출되었습니다. 모델링된 IA 쥐에게 혈관 내피 손상을 감지하기 위해 침묵하거나 과발현된 MALAT1을 주사했습니다. IA 환자의 혈관 내피 세포를 추상화하고 침묵 또는 과발현된 MALAT1으로 형질감염시켜 MALAT1이 세포 생존 및 세포자멸사에 미치는 영향을 확인했습니다. MALAT1, miR-143 및 VEGFA 간의 연결은 온라인 예측, 루시퍼라제 활성 및 RNA-풀다운 분석에 의해 확인되었습니다. IA 조직에서 MALAT1 및 VEGFA의 과발현 및 miR-143의 불량한 발현이 발견되었습니다. MALAT1의 하향 조절은 혈압, ET-1, vWF 및 MMP-9의 발현을 억제했을 뿐만 아니라 IA가 있는 쥐의 혈관 내피 세포의 세포 사멸 지수를 억제했습니다. 하향 조절된 MALAT1은 IA에서 세포 사멸을 억제하고 혈관 내피 세포의 생존을 촉진했습니다. MALAT1은 miR-143 및 miR-143에 결합하여 VEGFA를 표적화했습니다. 이 연구는 MALAT1이 miR-143에 대한 경쟁적 결합을 통해 VEGFA 발현을 증가시켜 IA에서 세포자멸사를 촉진하고 혈관 내피 세포의 생존력을 약화시킨다는 것을 시사합니다.
뇌동맥류라고도 알려진 두개내 동맥류(IA)는 뇌동맥강 또는 동맥벽의 국소적 비정상적 확장에 의해 유발되는 두개내압의 증가에 의해 유발된다[1]. IA는 사망률과 이환율이 높은 중증 질환으로, 유병률은 일반 인구의 약 1~3%이다[2]. IA의 주요 임상적 특징은 뇌혈관경련, 자발성 뇌출혈, 안구운동신경마비이다[3]. 지금까지 IA의 발생과 발달을 이끄는 일반적인 위험 인자는 혈역학적 장애, 유전자, 노화, 감염 및 선천적 요인을 포함한다[4]. 주로 외과적 클리핑 및/또는 혈관내 코일링과 같은 주요 임상 치료법은 동맥류 파열을 예방하는 기능을 합니다[5]. 그러나 IA를 관리하는 더 효과적인 방법에 대한 시급한 필요성을 반영하여 IA의 기본 메커니즘은 여전히 해명되어야 합니다.
긴 비암호화 RNA(lncRNA)는 비암호화 RNA 종에 속하는 200개 이상의 뉴클레오티드입니다[6]. IA에서 lncRNA 성장 정지 관련 lncRNA 1의 발현은 하향조절되는 것으로 보고되어 있다(MALAT1)는 길이가 약 8000 nt인 고도로 농축되고 널리 발현되는 lncRNA이다[7]. MALAT1은 흉부 대동맥류에서 평활근 기능 장애를 조절하는 것으로 문서화되었습니다[8]. 또한, IA에서 lncRNA와 mRNA(mRNA)의 비정상적 발현이 보고되었으며, lncRNA-mRNA 동시발현 네트워크가 IA의 발병기전을 찾는 단서를 제공한다[9]. MALAT1은 인간 골수 중간엽 줄기 세포에서 miRNA-143(miR-143) 발현을 표적으로 하여 오스테릭스 발현을 촉진하고 골형성 분화를 조절하는 것으로 제안되었습니다[10]. 한 연구는 또한 평활근 세포에서 krüppel-like factor 5의 조절과 IA에서 수축성 및 증식을 역전시키는 miR-143/145 클러스터의 역할을 제시했습니다[11]. Feng et al.에 따르면 순환 중 miR-143/145 및 더 높은 MMP-9 수준의 하향 조절은 IA의 형성 및 파열과 관련이 있을 수 있습니다[12]. 분석에 따르면 대부분의 제어되지 않는 miRNA(miR-143 및 miR-145)는 혈관 내피 성장 인자(VEGF)와 같은 신호 경로인 표적 유전자 및 세포 이동 또는 부착을 조절하는 기타 유전자에 공통적임이 밝혀졌습니다[13] . 한 연구에서 IA에서 혈관 내피 성장 인자-A(VEGFA) 변이의 예측 중요성이 밝혀졌습니다[14]. 따라서 우리는 혈관 내피 손상에 의해 유도된 IA에서 MALAT1/miR-143/VEGFA 신호 축의 메커니즘을 평가하고자 했습니다.
이 연구는 USTC 제1부속 병원의 기관 검토 위원회, 중국 과학 기술 대학교 생명 과학 및 의학과의 승인을 받았으며 헬싱키 선언의 신조를 따랐습니다. 참가자는 이 연구에서 서면 동의서를 제공했습니다. 모든 동물 실험은 국립 보건원의 실험 동물 관리 및 사용에 대한 지침에 따라 수행되었습니다. 프로토콜은 중국 과학 기술 대학교 생명 과학 및 의학부 USTC 제1 부속 병원 동물 실험 윤리 위원회에서 승인했습니다.
USTC 제1부속병원, 중국과학기술대학교 생명과학 및 의학부에서 영상 검사로 진단되어 신경외과 클리핑을 받은 20명의 IA 환자(IA 그룹)가 실험에 선택되었습니다. 43.27±6.25 세의 남자가 11명, 여자가 9명이었다. IA 조직을 샘플링했습니다. 한편, 편도체와 해마 경화증에 의한 측두엽 간질 환자 20명(대조군)에서 측두엽의 측두피질동맥혈관조직을 절제하였다. 절제된 조직은 수술 후 조직병리학적 검사를 통해 정상 동맥조직으로 확인되었으며, 나이 44.18±5.91 세는 남자 13명, 여자 7명이었다. IA 그룹과 대조군(둘 다 P> 0.05). 공복 상태의 모든 피험자로부터 수술 전 아침 같은 시간에 정맥혈 샘플(2개 튜브)을 채취했습니다.
60마리의 깨끗한 등급 Sprague-Dawley 수컷 쥐(Hunan SJA Laboratory Animal Co., Ltd., Hunan, China), 체중 200~250 g, 7일(25 ± 2 °C, 상대 습도 65~70) %, 12 h의 명암 주기, 자유 물 및 음식 섭취). 쥐를 돼지 췌장 엘라스타제와 함께 외부 경동맥과 분기 동맥 벽 주위에 떨어뜨렸습니다. 외경동맥은 약 1.5 mm의 외경동맥 가지에서 2개의 수술선으로 결찰되었다. 외부 경동맥은 두 라인 사이에서 절단되어 외부 경동맥의 블라인드 분절에 내부 경동맥류를 형성했습니다. 쥐에게 수술 후 1주일 동안 1% 식염수를 먹였습니다. 1 개월 후에 뇌혈관조영술을 시행하였고 동맥류의 형성이 관찰되었다.
IA 쥐 모델을 설정한 후 50마리의 쥐를 무작위로 빈 그룹(n =10, 모델링된 쥐는 100μL 인산완충식염수(PBS)의 정위주사를 하루에 한 번), 짧은 헤어핀 RNA(sh)-음성 대조군(NC) 그룹(n =10, 모델링된 쥐는 100 μL sh-MALAT1 NC의 정위 주사로 하루에 한 번), sh-MALAT1 그룹(n =10, 모델링된 랫트에 100μL sh-MALAT1 플라스미드를 1일 1회 정위 주사로 처리함), 과발현(Oe)-NC 그룹(n =10, 모델링된 쥐는 100 μL Oe-MALAT1 NC 플라스미드를 1일 1회 정위주사하고 Oe-MALAT1 그룹(n =10, 모델링된 쥐는 하루에 한 번 100 μL Oe-MALAT1 플라스미드의 정위 주사로 처리되었습니다[15]. 위의 플라스미드는 Shanghai Genechem Co., Ltd.(Shanghai, China)에서 구성되었습니다.
쥐 꼬리동맥의 혈압은 수술 후 1주, 4주, 12주에 측정하였다. 혈압을 측정하기 전에 외부 온도의 교란을 방지하기 위해 쥐를 항온 가열 장치에 잠시 두었다. 두 번째로, 쥐는 활동의 방해를 방지하기 위해 특수 쥐 케이지에서 몇 분 동안 조용하게 유지되었습니다. 혈압이 크게 변하는 경우에는 평균값을 얻기 위해 서로 다른 시간에 2~3회 측정하였다.
3 개월 후 쥐에게 1% pentobarbital sodium(40 mg/kg)을 복강 주사하여 마취시켜 정맥에서 혈액 샘플을 채취했습니다. 쥐를 안락사시키고 가슴을 열고 좌심실을 대동맥으로 삽관하고 대동맥을 절단하여 혈액을 방출했습니다. 한편, 헤파린나트륨이 함유된 생리식염수 30 mL를 이용하여 급속심장관류로 혈액을 플러싱한 후, 10% polyformaldehyde/0.1 M PBS(pH 7.4) 10 mL를 뇌에 주입하였다. 관류 및 고정 후 쥐의 뇌를 열었습니다. 두개골 기저부의 동맥 순환을 주의 깊게 관찰하고, 동맥류 조직을 분리하고, 현미경으로 동맥류의 변화를 관찰했습니다.
혈청 관련 지수는 ELISA 키트로 테스트했습니다. 채혈한 혈액 샘플을 37 °C 항온조에 1 시간 동안 두고 3000 r/min의 속도로 10분 동안 원심분리했습니다. 엔도텔린-1(ET-1) 및 폰 빌레브란트 인자(vWF) 발현의 검출은 키트의 지침에 따라 수행되었습니다(모든 키트는 중국 강소에 있는 NanJing JianCheng Bioengineering Institute에서 구입했습니다).
샘플을 10% 포르말린으로 24시간 이상 고정하고 파라핀 블록에 보존했습니다. 파라핀 블록을 자일렌으로 20분 동안 탈랍하고, 알코올(100%, 95%, 80%, 75%)의 기울기 내림차순으로 1분 동안 탈수하고, 헤마톡실린으로 10분 동안 염색했습니다. 그런 다음 조직을 증류수로 헹구고 염산 에탄올로 30초 동안 분별하고 50°C의 따뜻한 물에 5분 동안 담가두었습니다. 에오신 용액으로 염색한 조직을 증류수로 헹구고 70%, 90% 알코올로 탈수하고 자일렌으로 제거하고 중성검으로 밀봉하였다. 조직의 형태는 고배율 현미경으로 관찰되었습니다.
여분의 조직은 2.5% glutaric dialdehyde와 1% osmitic acid로 고정하고 탈수 후 Epon812 resin으로 포매하였다. 반박편은 톨루엔블루로 염색하고 다듬어서 초박편으로 만들었다. 섹션은 uranyl acetate와 lead citrate로 염색하고 JME-2000EX 투과 전자 현미경(Hitachi High-Technologies Co., Ltd., Shanghai, China)으로 관찰했습니다.
TUNEL 염색은 TUNEL 키트(Roche, Basel, Switzerland)를 기반으로 IA 쥐의 내피 세포 세포 사멸을 관찰하는 것으로 암시되었습니다. 준비된 쥐의 동맥류 절편을 자일렌으로 2회(5분/회) 세척하고 알코올의 내림차순(100%, 95%, 80%, 75%)으로 3회(5분/회) 탈수하였다. 조직을 DNase가 없는 20 μg/mL protease K 용액으로 15-30분 동안 처리하고, 50μL TUNEL 반응 용액을 60분 동안 적하하고, 50μL diaminobenzidine(DAB)으로 25 °C에서 10분 동안 현상했습니다. 그런 다음, 섹션을 헤마톡실린으로 대조염색하고, 구배 알코올로 탈수하고, 자일렌으로 제거하고, 중성 검으로 밀봉했습니다. 절편을 광학현미경으로 관찰하고 세포사멸 지수를 계산했습니다.
내피세포 분리는 Boscolo et al. [16]. IA 조직은 3 mm 2 로 절단되었습니다. 파편. 조직을 37°C에서 25분 동안 0.1% 콜라게나제 B/0.1% 디스파제(Roche)와 함께 인큐베이션했습니다. 그런 다음 미리 분리된 조직을 2 mL 피펫으로 2분 동안 분쇄하고 100μm 스트레이너(Thermo Fisher Scientific, Rockford, IL, USA)로 여과했습니다. 이어서, 세포 현탁액을 원심분리한 다음 MV2 배지(성장 인자 및 20% 소 태아 혈청 포함)(PromoCell, Heidelberg, Germany)에 재현탁시켰다. 세포를 1 × 10 4 로 시딩했습니다. 1 μg/cm 2 로 코팅된 배양 플라스크의 cells/mL 피브로넥틴. Jackson et al.에 의해 설명된 방법을 따릅니다. [17], Ulex europaeus Agglutinin I (UEA) (Vector Laboratories, Ltd., Peterborough, UK)로 코팅된 비드 (Dynabeads M-450 Tosylactived, Oxoid, Hampshire, UK)로 80-100% confluence의 세포를 분리했습니다. . 렉틴이 코팅된 비드에 부착된 내피세포를 자성입자 농축기로 집적하고, 비접합 세포를 기본 배지로 세척하였다. UEA 양성 세포를 배양 배지에 재현탁하고 피브로넥틴이 코팅된 배양 플라스크에 시딩하여 세포의 접착력 및 성장 속도를 개선했습니다.
세포는 피브로넥틴 코팅된 챔버 슬라이드에서 MV2에서 성장되었습니다. 세포 융합이 80-100%에 도달하면 세포를 4 °C에서 아세톤에 고정하고 1% Triton X-100으로 5분 동안 처리한 다음 0.5% 소 혈청 알부민(BSA)으로 15분 동안 처리했습니다. 세포에 vWF에 대한 1차 항체(1:300, Abcam, Cambridge, MA, USA)를 적하하고 2시간 동안 인큐베이션하고(일차 항체 없이 NC를 수행함), 양고추냉이 퍼옥시다제-추정 면역글로불린 G(1:150, Abcam) 및 30분 동안 인큐베이션했습니다. 그런 다음 세포를 25 °C에서 5분 동안 50 L DAB로 발달시키고, 헤마톡실린으로 대조염색하고, 0.1% 염산으로 분화하고, 알코올로 탈수하고, 크실렌 제거 및 중성 검 밀봉을 수행했습니다. 건조 후 세포를 도립현미경으로 촬영하였다.
대수기의 동맥류 혈관 내피 세포는 5개 그룹으로 할당되었습니다:빈 그룹(처리하지 않은 동맥류 혈관 내피 세포), sh-NC 그룹(sh-MALAT1 NC 플라스미드로 형질감염된 동맥류 혈관 내피 세포), sh-MALAT1 그룹( sh-MALAT1 플라스미드로 형질감염된 동맥류 혈관 내피 세포), Oe-NC 그룹(Oe-MALAT1 NC 플라스미드로 형질감염된 동맥류 혈관 내피 세포), 및 Oe-MALAT1 그룹(Oe-MALAT1로 형질감염된 동맥류 혈관 내피 세포). 상기 플라스미드는 Genechem에 의해 합성되었습니다. 리포펙타민 TM 지침에 따라 세포 형질감염을 수행했습니다. 2000 시약(11668-027, Invitrogen, Carlsbad, California, USA).
각 그룹의 혈관 내피 세포를 96웰 플레이트에 3 × 10 4 밀도로 파종했습니다. 세포/mL 및 37 °C, 5% CO2에서 배양 48 시간 동안. 각 그룹은 5개의 평행 웰로 설정되었고 각 웰에는 20 μL의 신선한 MTT 용액(5 mg/mL, Sigma, St. Louis, MO, USA)이 추가되었습니다. 4시간 반응 후, 세포를 200μL 디메틸 설폭사이드와 혼합하였다. 완전히 용해된 후 각 그룹의 세포의 광학 밀도 값은 490 nm에서 microplate reader(BioRad, Hercules, California, USA)로 측정되었습니다.
세포 주기 분포는 요오드화 프로피디움(PI) 염색으로 테스트되었습니다. 혈관 내피 세포를 분리하고 원심분리하고 미리 냉각된 75% 에탄올로 재현탁하고 -20 °C에서 밤새 고정했습니다. 세포를 원심분리하여 상층액을 버렸다. 세포를 450μL PBS에 추가하고, 100μL Rnase A를 첨가하고, 빛을 피하면서 30분 동안 4°C에서 400μL PI로 염색했습니다. 유세포 분석기(FACSCalibur, Becton, Dickinson and Company, NJ, USA)를 사용하여 세포 주기 분포를 테스트했습니다.
세포 사멸은 Annexin V/PI 이중 염색에 의해 테스트되었습니다. 분리된 내피세포를 모아 PBS로 3회 세척하였다. 세포를 100 μL 미리 냉각된 1x 결합 완충액으로 재현탁하고 각각 5 μL Annexin 및 5 μL PI와 혼합했습니다. 세포 사멸은 유세포 분석기로 테스트되었습니다. AnnexinV를 가로축으로, PI를 세로축으로 하여 왼쪽 상부 사분면은 기계적 손상 세포를 나타냅니다(AnnexinV - /PI + ), 후기 아폽토시스 세포 또는 괴사 세포에 대한 우측 상부 사분면(AnnexinV + /PI + ), 음성 정상 세포에 대한 왼쪽 하단 사분면(AnnexinV - /PI − ), 초기 세포사멸 세포에 대한 오른쪽 하단 사분면(AnnexinV + /PI − ). 총 아폽토시스 세포(AnnexinV + /PI − 및 AnnexinV + /PI + )를 계산하여 백분율로 표시했습니다.
조직과 세포의 총 RNA를 Trizol(Takara Biotechnology Ltd., Dalian, China)로 추출하고 RNA의 농도와 순도를 측정했습니다. RNA의 상보적 DNA로의 역전사 과정은 역전사 키트(K1621, Fermentas, Maryland, NY, USA)의 지침에 따라 수행하였다. MALAT1, miR-143 및 VEGFA 프라이머 서열(표 1)은 Genechem에서 고안하고 구성했습니다. lncRNA, miRNA 또는 mRNA의 발현을 평가하기 위해 Roche Real-Time PCR 시스템(Roche)과 함께 SYBR GreenPCR Master Mix(Takara, Tokyo, Japan)를 사용하여 RT-qPCR을 수행했습니다. U6은 miR-143의 내부 매개변수로 설정되었으며 MALAT1 및 VEGFA는 GAPDH(glyceraldehyde-3-phosphate dehydrogenase)를 내부 매개변수로 사용했습니다. 타겟 유전자의 상대적 전사 수준은 2 -△△Ct 로 계산되었습니다. 방법.
조직 및 세포의 총 단백질을 추출했습니다. 단백질 농도는 비신코닌산 키트(Boster Biological Technology Co. Ltd., 무한, 후베이, 중국)의 지침에 따라 결정되었습니다. 단백질은 10% 폴리아크릴아미드 겔(Boster Biological Technology)을 사용한 전기영동에 의해 분리되었습니다. 멤브레인을 폴리비닐리덴 플루오라이드 멤브레인으로 옮기고 1 시간 동안 5% BSA로 밀봉했습니다. 멤브레인은 Ki-67(1:1000), VEGFA(1:1000), vWF(1:1000) 및 매트릭스 메탈로프로테이나제(MMP)-9(1:1000, Abcam, Cambridge, UK)에 대한 1차 항체와 함께 배양되었습니다. , Cyclin D1(1:1000), Bax(1:1000) 및 Bcl-2(1:1000, Santa Cruz Biotechnology, Santa Cruz, California, USA) 및 GAPDH(1:2000, Jackson Immuno Research, Grove, Pennsylvania, USA) 및 양고추냉이 과산화물로 표지된 2차 항체(1:500, Jackson Immuno Research)로. 멤브레인은 Odyssey 2색 적외선 형광 스캐닝 이미징 시스템으로 얻었고 밴드의 회색 값은 Quantity One 이미지 분석 소프트웨어로 측정했습니다.
MALAT1 및 miR-143의 결합 부위는 생물정보학 웹사이트(https://cm.jefferson.edu/rna22/Precomputed/)에 의해 예측 및 설명되었습니다. MALAT1과 miR-143 사이의 결합 관계는 루시퍼라제 리포터 유전자 분석에 의해 추가로 확인되었습니다. 합성 MALAT1 3' 비번역 영역(3'UTR) 유전자 단편을 pmiR-Report 루시퍼라제 리포터 벡터(Thermo Fisher Scientific)에 도입하여 엔도뉴클레아제 부위 Bamh1 및 Eco1에 의해 MALAT1 야생형(MALAT1-WT)을 생성했습니다. MALAT1-WT 상에서 서열의 상보적 서열 돌연변이 부위를 고안하고, 표적 단편을 pmiR-Report 루시퍼라제 리포터 벡터에 삽입하여 제한 엔도뉴클레아제 분해 후 T4 DNA 리가제에 의해 MALAT1 돌연변이형(MALAT1-MUT)을 생성하였다. 올바르게 시퀀싱된 MALAT1-WT 및 MALAT1-MUT를 모방 NC 및 miR-143 모방으로 혈관 내피 세포에 공동 형질감염시켰다. 형질감염 48시간 후에 세포를 수확하고 용해하고, 루시퍼라제 활성을 발광계(Glomax20/20, Promega, Madison, Wisconsin, USA)가 있는 루시퍼라제 검출 키트(BioVision, San Francisco, CA, USA)로 테스트했습니다.
miR-143과 VEGFA의 표적 관계 및 miR-143과 VEGFA 3'UTR의 결합 부위는 생물정보학 웹사이트(http://www.targetscan.org/vert_72/)를 통해 예측하였다. miR-143 결합 부위를 포함하는 VEGFA 3'UTR 프로모터 영역의 서열을 합성하고 pmiR-Report 루시퍼라제 리포터 벡터에 클로닝하여 VEGFA-WT를 생성했습니다. 이 리포터를 기반으로 VEGFA-WT와 miR-143의 결합 부위가 돌연변이되어 VEGFA-MUT를 형성했습니다. VEGFA-WT 또는 VEGFA-MUT 리포터를 모방 NC 또는 miR-143 모방과 혼합한 다음 48시간 동안 혈관 내피 세포에 공동 형질감염시켰다. 그 후, 세포를 용해하고 루시퍼라제 검출 키트로 루시퍼라제 활성을 테스트했습니다.
miR-143과 MALAT1 간의 결합 관계를 확인하기 위해 RNA-풀다운 분석을 구현했습니다. 3개의 비오틴 표지 miRNA 서열 Bio-miR-143-WT, Bio-miR-143-MUT 및 Bio-miR-NC를 설계하고 GenePharma Company(중국 상하이)에 위탁했습니다. 이러한 비오틴화된 올리고뉴클레오티드를 48시간 동안 세포에 형질감염시켰다. 그런 다음, 세포를 수확하고 10분 동안 특정 세포 용해물(Ambion, Austin, Texas, USA)과 함께 인큐베이션했습니다. 그 후, lysate를 RNase-free 및 효모 tRNA(모두 Sigma)로 미리 코팅한 M-280 streptavidin 비드로 4 °C에서 3 시간 동안 부화한 다음, 차가운 용해액으로 두 번, 낮은 온도로 세 번 세척했습니다. 염 완충액, 고염 완충액으로 1회. 길항 miR-143 프로브가 NC로 설정되었습니다. 총 RNA는 Trizol로 추출한 다음 RT-qPCR을 사용하여 MALAT1 농축 수준을 테스트했습니다.
모든 데이터는 SPSS 21.0 소프트웨어(IBM Corp. Armonk, NY, USA)에 의해 설명되었습니다. 측정 데이터는 평균±표준편차로 나타내었다. 두 그룹 간의 비교는 독립 표본 t에 의해 수행되었습니다. 여러 그룹 간의 비교는 일원 분산 분석(ANOVA)으로 평가하고 쌍별 비교는 Tukey의 다중 비교 테스트로 구현했습니다. MALAT1 발현과 IA의 임상병리학적 특징 사이의 관계는 카이-제곱 검정에 의해 결정되었다. 피 0.05 미만의 값은 통계적으로 유의한 차이를 나타냅니다.
<섹션 데이터-제목="결과">IA군과 대조군의 혈청 내 ET-1 및 vWF 발현을 ELISA로 검출하였으며, 그 결과 대조군에 비해 IA군에서 ET-1 및 vWF의 발현이 증가하는 것으로 나타났다(P <0.05) (그림 1a, b).
<그림>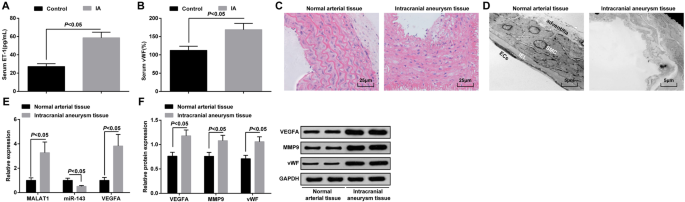
MALAT1 및 VEGFA는 과발현되고 miR-143은 IA 조직에서 하향 조절됩니다. 아 ELISA에 의한 IA 환자 및 측두엽 간질 환자의 혈청에서의 ET-1 발현. ㄴ ELISA에 의한 IA 환자 및 측두엽 간질 환자의 혈청에서의 vWF 발현. ㄷ H 염색에 의한 IA 조직 및 정상 동맥 조직의 병리학적 관찰. d 투과전자현미경에 의한 IA 조직 및 정상 동맥 조직의 형태 관찰. 이 RT-qPCR에 의한 IA 조직 및 정상 동맥 조직에서의 MALAT1, miR-143 및 VEGFA mRNA 발현. 에 웨스턴 블롯 분석에 의한 IA 조직 및 정상 동맥 조직에서의 VEGFA, MMP-9 및 vWF 단백질 발현. 내피 세포(EC), 내부 탄성 판(IEL), 평활근 세포(SMC). 측정 데이터는 평균 ± 표준 편차로 표시되었습니다. 그룹 간의 비교는 독립 표본 t에 의해 수행되었습니다. 테스트
H 염색을 통해 IA 조직의 병리학적 변화를 관찰하였다. 정상 동맥 조직에서는 내배엽, 내피세포, 평활근 세포에 뚜렷한 손상이 없었고 세포가 가지런히 배열되어 완전한 구조를 가졌다. IA 조직은 손상된 내피 세포, 퇴화된 평활근 세포, 약화된 동맥벽, 파열된 탄성 섬유 및 침윤된 염증 세포를 나타냈다(그림 1c).
투과전자현미경으로 IA 및 정상 동맥 조직의 미세구조 변화를 관찰하였다. 내피 세포가 완전하고 외막 구조가 손상되지 않은 것으로 밝혀졌습니다. 정상적인 동맥 조직에서는 부러진 내부 탄성막이나 세포자멸사 평활근 세포가 관찰되지 않았습니다. IA 조직에서는 대부분의 내피세포의 소실, 내부탄성층이 심하게 파손되어 평활근세포가 심하게 손상되고 퇴화되며, 혈관외막의 이상으로 주로 나타나는 혈관벽의 심각한 퇴행성으로 나타났다. (그림 1d).
RT-qPCR은 MALAT1, miR-143 및 VEGFA mRNA 발현을 결정하기 위해 수행되었으며, IA 조직 및 정상 동맥 조직에서 VEGFA, MMP-9 및 vWF 단백질 발현에 대한 웨스턴 블롯 분석을 수행했습니다. 정상 동맥 조직과 대조적으로 MALAT1, VEGFA, MMP-9 및 vWF 발현 수준이 IA 조직에서 증가하고 miR-143 발현이 저하됨이 입증되었습니다(모든 P <0.05) (그림 1e, f).
MALAT1의 중앙값 발현에 비추어 환자를 저발현 그룹과 고발현 그룹의 두 그룹으로 할당했습니다. IA 환자의 MALAT1 발현과 임상병리학적 특징 사이의 관계를 분석하였다. 결과는 Hunt-Hess 등급, 내피 손상 정도 및 흡연 이력이 MALAT1 발현과 관련이 있음을 시사했습니다(모두 P <0.05), 연령, 성별 및 수술 모드는 MALAT1 발현과 관련이 없었습니다(모두 P> 0.05) (표 2).
표 3에 나타난 바와 같이 MALAT1 knockdown은 저하된 반면 MALAT1 복원은 4주차와 12주차에 혈압을 높였습니다.
ELISA는 MALAT1 하향 조절이 감소한 반면 MALAT1 상향 조절은 IA가 있는 쥐의 혈청에서 ET-1 및 vWF 발현을 증가시키는 것으로 나타났습니다(그림 2a, b).
<그림>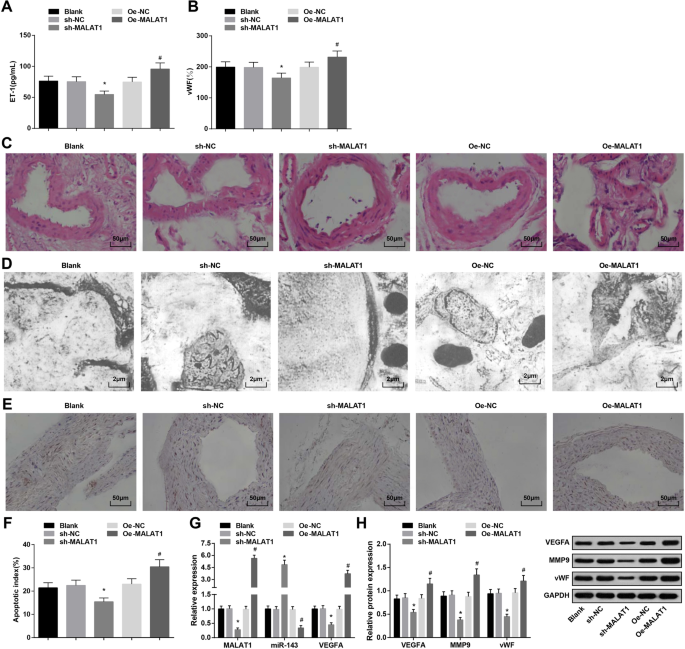
하향 조절된 MALAT1은 혈압, ET-1, vWF 및 MMP-9의 발현뿐만 아니라 IA가 있는 쥐의 혈관 내피 세포의 세포 사멸 지수를 억제합니다. 아 ELISA에 의한 쥐의 혈청에서의 ET-1 발현. ㄴ ELISA에 의한 쥐의 혈청에서의 vWF 발현. ㄷ HE 염색으로 관찰된 쥐에서 IA 조직의 병리학적 변화. d 투과전자현미경으로 관찰한 쥐의 IA 조직의 미세구조. 이 TUNEL 염색에 의한 혈관 내피 세포의 아폽토시스. 에 쥐의 혈관 내피 세포 사멸 지수. 지 RT-qPCR에 의한 쥐의 IA 조직에서 MALAT1, miR-143 및 VEGFA mRNA 발현. 아 웨스턴 블롯 분석에 의한 쥐의 IA 조직에서 VEGFA, MMP-9 및 vWF 단백질 발현. * 피 <0.05 vs. sh-NC 그룹, # P <0.05 대 Oe-NC 그룹. 측정 데이터는 평균 ± 표준 편차로 표시되었으며, 여러 그룹 간의 비교는 Tukey의 다중 비교 테스트에 따른 일원 분산 분석으로 평가되었습니다.
각 군의 IA 조직의 병리학적 변화를 H 염색으로 조사하였다. 블랭크군, sh-NC군, Oe-NC군은 내막손상, 내피세포 박리, 평활근세포 퇴화, 세포와 층 감소, 동맥벽 얇아짐을 보였다. sh-MALAT1 군에서 두개내동맥의 내피, 내피세포, 평활근세포층, 외막층이 약간 손상되었으나 깔끔하게 배열되었다. Oe-MALAT1 군은 내막이 사라지고 내피세포가 박리되고 탄력섬유가 끊어지고 염증세포가 침윤된 모습을 보였다(Fig. 2c).
각 군의 쥐의 IA 조직을 투과전자현미경으로 관찰하였다. 블랭크군, sh-NC군, Oe-NC군에서 내피세포 변성, 내피하층 붕괴, 내부탄성층 소실, 평활근세포 감소가 나타났다. sh-MALAT1 그룹은 편평한 내피 세포, 타원형 핵 및 증가된 콜라겐 섬유를 나타내었지만 탄성 층이 없는 것으로 나타났습니다. Oe-MALAT1 그룹은 내피 세포가 사라지고 근육층에서 탄성층이 분리되어 내강으로 분해되는 특징이 있습니다(그림 2d).
IA 쥐의 혈관 내피 세포의 세포 사멸 지수를 TUNEL 염색으로 테스트했습니다. MALAT1을 억제하면 혈관 내피 세포의 세포 사멸 지수가 감소한 반면 MALAT1이 과발현되면 반대 효과를 나타냅니다(그림 2e, f).
MALAT1, miR-143 및 VEGFA mRNA 발현의 RT-qPCR 검출 및 IA 조직에서 VEGFA, MMP-9 및 vWF 단백질 발현의 웨스턴 블롯 분석은 MALAT1 제거가 MALAT1, VEGFA, MMP-9 및 vWF 발현을 억제한다는 것을 제시했습니다 , 그리고 miR-143 발현을 높였습니다. 반대로, MALAT1 상승은 이러한 유전자 발현에 반대의 영향을 주었다(그림 2g, h).
면역조직화학적 염색을 이용하여 내피 특이적 마커 vWF의 발현을 검출하였다. IA 혈관 내피 세포의 세포질은 미세 갈색 입자로 덮여 있는 것으로 나타났으며 이는 양성인 반면 NC 그룹의 세포질에는 갈색 입자가 없었습니다. 그 결과 배양된 세포가 내피세포임을 확인하였다(Fig. 3a, b).
<그림>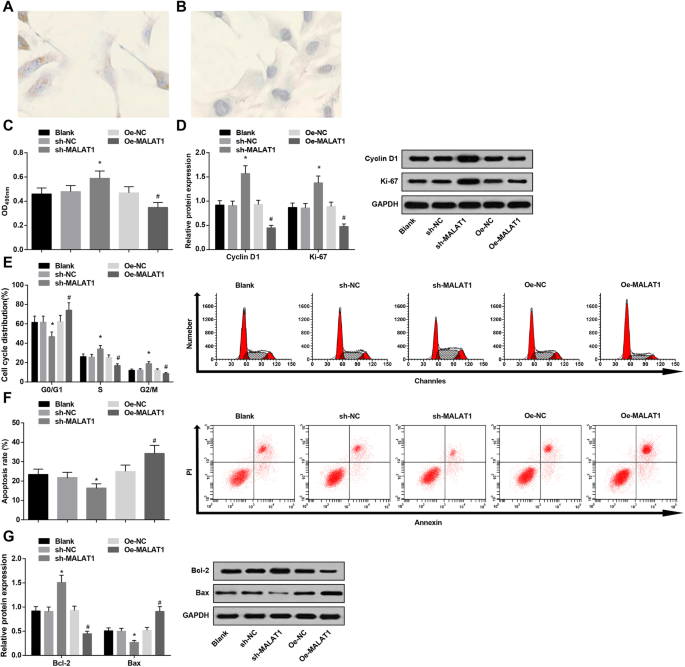
Low expression of MALAT1 advances viability and restrains apoptosis of vascular endothelial cells in IA. 아 vWF immunohistochemical staining in IA vascular endothelial cells:IA vascular endothelial cells were covered with fine yellow particles. ㄴ vWF immunohistochemical staining in IA vascular endothelial cells:IA vascular endothelial cells showed no brown particles in the NC group. ㄷ Vascular endothelial cell viability in each group by MTT assay. d Protein expression of CyclinD1 and Ki-67 in each group by western blot analysis. 이 Cell cycle changes in each group by PI staining. 에 Cell apoptosis rate in each group by Annexin V/PI double staining. 지 Bax and Bcl-2 protein expression in each group by western blot analysis. * P <0.05 vs. the sh-NC group, # P <0.05 vs. the Oe-NC group. Measurement data were depicted as mean ± standard deviation, and comparisons among multiple groups were assessed by one-way analysis of variance followed with Tukey’s multiple comparisons test
MTT assay, flow cytometry, together with western blot analysis were utilized to test vascular endothelial cell viability and apoptosis. It was displayed that MALAT1 diminution promoted vascular endothelial cell viability (heightened Cyclin D1 and Ki-67 expression) and depressed apoptosis (decreased G0/G1 phase cells and increased S and G2/M phase cells, reduced Bax and elevated Bcl-2 expression). However, MALAT1 upregulation functioned in an opposite way to that of MALAT1 diminution on cell viability and apoptosis (Fig. 3c–g).
MALA1, miR-143, and VEGFA expression in vascular endothelial cells of each group were verified through RT-qPCR and western blot analysis. The results presented that MALA1 knockdown depressed MALA1 and VEGFA expression and enhanced miR-143 expression. On the contrary, MALA1 upregulation led to the increment in MALAT1 and VEGFA expression and the reduction in miR-143 expression (Fig. 4a, b).

MiR-143 is bound to MALAT1 and VEGFA is a target gene of miR-143. 아 MALAT1, miR-143, and VEGFA mRNA expression in vascular endothelial cells of aneurysm in each group. ㄴ VEGFA protein expression in vascular endothelial cells of aneurysm in each group. ㄷ The binding sites of MALAT1 and miR-143 predicted by the bioinformatics website. d The regulatory relation of MALLA1 and miR-143 validated by dual luciferase reporter gene assay. 이 The binding relationship between MALAT1 and miR-143 verified by RNA-pull down assay. 에 The binding sites of miR-143 and VEGFA predicted by the bioinformatics website. 지 The regulatory relation of miR-143 and VEGFA validated by dual luciferase reporter gene assay. * P <0.05 vs. the sh-NC group, # P <0.05 vs. the Oe-NC group. Measurement data were depicted as mean ± standard deviation, comparisons between two groups were assessed by independent sample t test, and comparisons among multiple groups were assessed by one-way analysis of variance followed with Tukey’s multiple comparisons test
The specific binding region between MALAT1 and miR-143 was divined by online analysis software (Fig. 4c). The results of dual luciferase reporter gene assay revealed that the luciferase activity was impaired in the MALAT1-WT + miR-143 mimic group versus the MALAT1-WT + mimic-NC group (P <0.05). However, there was no distinct difference in luciferase activity in the MALAT1-MUT + miR-143 mimic group relative to that in the MALAT1-MUT + mimic-NC group (P> 0.05), indicating that miR-143 was specifically bound to MALAT1 (Fig. 4d). The results of RNA-pull down assay reported that the enrichment level of MALAT1 in the Bio-miR-143-WT group was heightened compared to the Bio-miR-NC group (P <0.05), but there was no distinct difference in MALAT1 enrichment level in the Bio-miR-143-MUT group (P> 0.05) (Fig. 4e).
Bioinformatics software divined a targeted relationship between miR-143 and VEGFA (Fig. 4f). The results of luciferase activity showed that the relative luciferase activity repressed after VEGFA-WT and miR-143 mimic co-transfected into vascular endothelial cells (P <0.05). However, the relative luciferase activity of vascular endothelial cells was not affected by co-transfection of VEGFA-MUT and miR-143-mimic (P> 0.05) (Fig. 4g). It was indicated that VEGFA was a direct target gene of miR-143.
IA is a serious intracranial disease, which mainly leads to subarachnoid hemorrhage [18]. A previous study has demonstrated the involvements of lncRNA-related competitive endogenous RNA networks in IA [19]. Also, a recent study has provided a proof that functional polymorphism in miR-143/145 gene promoter region is connected with the risk of IA [20]. In a study conducted by Xu et al. , it is shown that overexpression of angiogenic factors, such as VEGFA, may be related to IA formation and rupture [21]. In order to explain the molecular mechanism of MALAT1 in IA, a series of assays have been conducted and the results revealed that IA induced by vascular endothelial injury was regulated by MALAT1/miR-143/VEGFA signal axis.
Firstly, our study has provided substantial evidence that MALAT1 and VEGFA are upregulated and miR-143 is downregulated in IA tissues. A recent study has presented that MALAT1 is one of the most upregulated lncRNAs in the process of cerebral ischemia [22]. Another study has presented that MALAT1 is upregulated in ovarian cancer cells and intends to participate in the processes of ovarian cancer cell apoptosis, migration, and proliferation [23]. A study concerning to the expression profile of unruptured and ruptured IA has demonstrated that the expression of angiogenic factors such as VEGFA is upregulated in ruptured aneurysm [21]. Moreover, a clinical study has presented that the miR-143/145 cluster of IA patients is downregulated compared to healthy subjects [11]. In addition, it is previously discussed that miR-143/145 takes part in various biological processes associated with aneurysm formation and is downregulated in patients with IA [20]. All these findings are more or less echoed with the previous exploration outcomes.
Except for the abovementioned findings, this study has also explored the functional role of MALAT1 in IA through gain-off-function and loss-of-function assays. It could be summarized that downregulation of MALAT1 reduced blood pressure, serum levels of ET-1, and vWF and MMP-9 expression in IA tissues. It has been suggested previously that downregulation of MALAT1 restrains the upregulation of the glucose-induced ET-1 transcription product [24]. Also, it is reported that ectopic MALAT1 expression is the inducer of vWF generation [25]. Another study has verified that the depletion of MALAT1 in bone marrow-derived macrophages inhibits the expression of MMP-9 [26].
Also, cell experiments have been conducted for further confirmation of the functions of MALAT1 in IA. The results have suggested that MALAT1 knockdown promoted vascular endothelial cell viability and depressed apoptosis in IA. Similarly, it has been documented that disturbance of MALAT1 improves aortic mural architecture and retards aneurysm growth [8]. Supplementary to our study finding, there is research highlighting that poor expression of MALAT1 induces apoptosis and restrains proliferation of acute myeloid leukemia cells [27]. Another study has also demonstrated the inhibitory effects of MALAT1 knockdown on proliferation of human osteoarthritis cartilage cells [28]. Besides that, a prior research generally confirms that downregulation of MALAT1 can induce apoptosis and attenuate the proliferation of glioma cells [29]. Moreover, low expression of MALAT1 induced by RNA interference promotes apoptosis and suppresses proliferation of multiple myeloma cells [30]. Collectively, these studies have explained the molecular mechanism of MALAT1 in IA to some extent.
In addition, this study has evidenced that miR-143 is bound to MALAT1 and VEGFA is a target gene of miR-143. Similarly, a paper contends that MALAT1 binds directly to miR-143 and suppresses its expression [10]. Zhu et al. have found that MALAT1 exerts its roles via interacting with miR-143 in cervical carcinoma cells [31]. It is further confirmed that MALAT1 indirectly modulate VEGFA via miR-200b-3p [32]. Moreover, another study has suggested that miR-143-3p mediates ZFPM2 effect on a number of protein targets in blood, including VEGFA [33]. Nevertheless, the interactions among MALAT1, miR-143, and VEGFA in IA have not been discussed and need further study.
From these results, it is clear that MALAT1 knockdown depresses apoptosis and promotes viability of vascular endothelial cells in IA by modulating miR-143/VEGFA axis. The co-expression network suggests the connection between MALAT1 and miR-143 with the involvement of VEGFA. The findings in this study partially disclose the pathogenesis of IA initiation and progression, and the studied targets may be a notably potential entry point to reveal pathology of IA from another perspective. Limitedly, further large-scale studies are required to comprehensively illustrate the mechanisms of MALAT1/miR-143/VEGFA axis in IA.
나노물질
CNC 라우터 기계를 구입할 생각이 있을 때 CNC 라우터 키트에 몇 개의 축이 필요합니까? 모든 CNC 라우터 구매자의 공통된 문제이므로 3축, 4축 및 5축 CNC 라우터 비교를 시작하겠습니다. CNC 라우터 머신 키트의 3축, 4축, 4축 및 5축 이해 5축:X-Y-Z-A-B, X-Y-Z-A-C, X-Y-Z-B-C (스핀들은 좌우 180도 회전할 수 있습니다.) 4축:X-Y-Z-A, X-Y-Z-B, X-Y-Z-C(4축 연결) 4축:Y-Z-A, X-Z-A(3축 연결) 3축:X-Y-Z(3축 연결) A, B
밀링은 CNC 정밀 가공에 사용되는 중요한 기술입니다. , 의료, 항공 우주, 광학 및 기계 부품에 적용됩니다. 밀링은 회전 도구를 사용하여 도구 축에 대해 비스듬히 공작물을 공급함으로써 공작물에서 재료를 제거합니다. 지침은 CAD 파일을 통해 CNC 공작 기계에 입력되고 일련의 정확한 시퀀스 지침으로 변환됩니다. CNC 공작 기계는 이러한 프로그래밍 명령을 사용하여 물리적 작업자 없이 자동으로 작동합니다. 제조업체는 비용 절감, 속도 향상, 정확도 향상 및 생산성 향상과 같은 CNC 가공의 적용을 통해 많은 이점을 얻었습니다.