이 작업에서 일련의 전이 금속(Cr, Mn, Fe 및 Co) 도핑된 카올리나이트 나노클레이가 밀도 기능 이론(DFT) 계산에 의해 조사되었습니다. 금속 도핑이 카올리나이트의 기하학적 구조와 전자 구조에 미치는 영향을 분석하였다. 강자성(FM), 반강자성(AFM) 및 비자성(NM) 상태의 전이 금속(TM) 도핑된 카올리나이트 구조가 연구되었습니다. 결정 부피, 격자 매개변수, 결합 길이, 전하 및 스핀은 분산 보정 밀도 기능 이론(DFT-D2)에 의해 계산되었습니다. 결과는 Cr
3+
및 Fe
3+
도펀트는 AFM 상태에서 더 안정적인 반면 Mn
3+
AFM 및 FM 상태 모두 선호, Co
3+
도펀트는 NM 상태를 선호합니다. 또한, 전이 금속 도핑은 격자 부피 팽창과 밴드 갭에서 일부 도펀트 상태를 유발할 수 있습니다.
<섹션 데이터-제목="배경">
배경
고령토계 나노점토 광물은 열수 변화 및/또는 풍화 과정의 결과로 층 구조, 작은 입자 크기, 가장 중요한 수산화 기가 많은 표면으로 인해 독특한 물리적 특성을 갖습니다. 그것은 재료 화학, 환경 화학 및 광물 물리학의 연구자들의 관심을 끌었다[1,2,3,4,5,6,7,8,9,10,11]. 지구상에서 가장 풍부한 나노점토 광물 중 하나인 카올리나이트는 플라스틱, 촉매 및 시멘트 산업에서 광범위하게 사용되었습니다. 새로운 지지체 재료로서 카올리나이트의 추가 기능화는 다양한 분야에서 점점 더 많은 관심을 끌고 있습니다. 카올리나이트는 단순히 다른 나노입자와 혼합하여 태양 에너지 유틸리티를 위한 상변화 물질을 형성하기 위한 지지 물질 역할을 하거나[4, 5], 도핑된 산화물로 코팅되어 전도성 분야에 적용하기 위한 전도성 분말을 형성할 수 있습니다[9, 12]. 카올리나이트와 기능성 나노입자의 혼성화는 시너지 효과를 통해 Pd-ZnO의 광촉매 활성과 CdS의 발광 특성을 향상시키는 것으로 밝혀졌다[6, 7]. 카올리나이트의 표면 특성은 표면에 일부 관능기를 고정하거나[13, 14] 추가 개선을 위한 산 활성화 전처리에 의해 수정되었습니다[2].
고령토계 광물의 구조와 에너지는 실험적으로[15,16,17] 이론적으로[18,19,20,21,22] 광범위하게 조사되었습니다. 카올리나이트 표면의 중금속 흡착에 대한 이론적 연구는 Cd, Cu, Hg 및 Ni(II) 흡착에 대해 연구되었으며[23], 이온에 대한 카올리나이트 점토의 흡착능은 Ni> Cu> Cd> Hg(II). 카올리나이트(001) 표면에서 Pb(II)[24, 25]와 uranyl[26]의 흡착 및 확산이 연구되었고[24,25,26], 수성 시스템에서의 흡착 거동도 나중에 보고되었다[27, 28]. 카올리나이트 표면에 대한 Mg, Ca 및 Fe 도핑의 영향 및 H2의 후속 흡착 및 침투 중간층으로의 O가 연구되었다[29]. H2의 흡착 에너지 도핑된 카올리나이트(001)의 O는 도핑되지 않은 표면보다 적게 발견되었습니다. 고유 결함이 있거나 없는 카올리나이트의 전자 구조는 표준 밀도 기능 이론(DFT) 기능 및 하이브리드 기능에 의해 연구되었습니다[30]. 그러나 최근까지 카올리나이트의 탈수산화, 탈알루미늄화 및 실리카 축합 과정 동안의 구조 진화가 DFT 계산에 의해 모델링되지 않았습니다[1, 31, 32]. 카올린계 재료에서 Al을 제거하면 이러한 층 재료의 기하학과 전자적 특성이 크게 바뀌고 지지 효과가 향상되었습니다[1, 2].
화합물의 구조와 특성을 수정하는 잘 알려진 방법인 금속 도핑은 Al2에 대해 이론적으로 연구되었습니다. O3 [33], TiO2 [34], MOF [35] 및 기타 고체 [36]. 전이 금속(TM) 도핑에 따른 카올리나이트 나노클레이의 구조 및 특성 변화를 조사하는 것은 이 층상 점토 재료에 대해 흥미로울 것입니다. 이 연구에서 일련의 Cr, Mn, Fe 및 Co 도핑된 카올리나이트 나노클레이가 DFT 계산에 의해 연구되었고 카올리나이트 나노클레이의 기하학적 구조 및 전자 구조에 대한 금속 도핑의 영향에 초점을 맞추었습니다. 이러한 전이 금속 도핑 카올리나이트 구조의 가능한 강자성(FM), 반강자성(AFM) 및 비자성(NM) 상태가 연구되었습니다. 격자 매개변수, 결합 길이, 전하 및 스핀은 분산 보정 밀도 기능 이론(DFT-D2)에 의해 최적화되고 계산되었습니다.
방법
모든 계산은 첫 번째 원칙 DFT를 기반으로 하는 프로그램 CASTEP(Cambridge Sequential Total Energy Package) 코드[37]로 수행되었습니다. Perdew, Burke 및 Ernzerhof(PBE)에 의한 교환 상관 가능성이 있는 일반화된 기울기 근사(GGA)가 계산에 사용되었습니다[38]. Grimme의 DFT-D2 분산 보정은 Van der Waals 분산 상호작용을 설명하기 위해 포함되었습니다[39]. 500 eV의 에너지 차단은 ultrasoft pseudo-potential 평면파 형식을 사용하여 적용되었습니다[40]. 2 × 2 × 3 k의 Monkhorst–Pack [41] 그리드 -점 메쉬는 기하학적 이완 및 전자 구조 계산에 사용되었습니다. 바닥 상태에서 일관된 전체 에너지는 밀도 혼합 방식에 의해 효과적으로 얻어졌습니다[42]. 지오메트리 최적화를 위해 SCF(Self-Consistent Field) 허용 오차에 대한 수렴 임계값은 1.0 × 10
−6
으로 설정되었습니다. eV/atom, 원자에 가해지는 모든 힘은 0.03 eV/Å 미만으로 수렴되었고, 총 응력 텐서는 0.05 GPa 정도까지 감소되었으며, 최대 이온 변위는 0.001 Å 이내였습니다. 원자가 상태에서 조사된 요소는 O(2s
2
2p
4
), Al(3s
2
3p
1
), Cr(3s
2
3p
6
3d
5
4초
1
), Mn(3d
5
4초
2
), Fe(3d
6
4초
2
) 및 Co(3d
7
4초
2
). Uspcc 의사 전위는 Mn, Fe 및 Co에 사용되었고 usp 의사 전위는 나머지 요소에 사용되었습니다. Broyden-Fletcher-Goldfarb-Shanno(BFGS) 최소화 알고리즘을 사용하여 형상 최적화 중에 셀 매개변수와 원자 조정이 완전히 완화되었습니다. 전자 접지 상태가 더 낮은 대칭을 채택할 수 있도록 TM 이온에 다른 초기 자기 모멘트를 부과하여 결정 대칭을 제거했습니다.
결과 및 토론
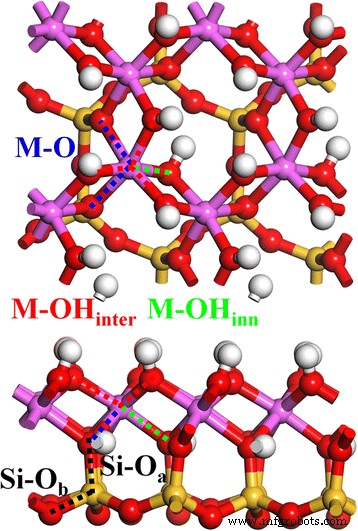
초기 카올리나이트 구조는 이전 작업 [1]에서 사용되었습니다. 그림 1은 카올리나이트(4 카올리나이트 단위)의 이완된 2 × 2 × 1 결정 구조를 보여줍니다. 카올리나이트 층 구조, Al2 시2 O5 (OH)4 , 8면체 Al-O 시트와 4면체 Si-O 시트로 구성되며 정점 O 원자(Oa ). Si-O 사면체는 중심 Si 원자 1개와 주변 O 원자 4개로 구성되며, 여기서 하나는 Oa입니다. 원자이고 나머지 세 개는 기본 O 원자입니다(Ob ). Al-O 팔면체는 중앙 Al 1개와 O 주변 6개로 구성되며, 여기서 2개는 Oa 원자와 나머지 4개는 다른 Al-O 팔면체와 공유되는 O 원자(OH 기에서)입니다. 게다가, 이러한 OH 그룹은 두 종류로 나눌 수 있습니다. 층간 OH(OHinter ) 층 구조의 표면과 내부 OH(OHinner ) Al 시트와 Si 시트 사이의 층 구조 내부. 따라서 Si-O 결합에는 두 가지 종류가 있습니다. Si-Oa 및 SiOb (검은색 점선), Al-O 결합 3종, Al-Ointer (빨간색 점선), Al–O내부 (녹색 점선) 및 Al–O(검은색 점선)는 카올리나이트 벌크 구조입니다.
분산 에너지는 층 사이의 상호 작용으로 인해 점토 광물의 구조 안정화에 항상 중요한 역할을 합니다[21, 43]. 여러 하이브리드 관능기 중 PBE-D2[21], B3LYP[22], B3LYP-D[18], RPBE-D2[18, 21]는 카올리나이트[44, 45]의 실험적 격자 구조를 얻는 데 사용되었습니다. ], PBE-D2 기능이 정확하고 시간이 덜 소요되는 것으로 나타났습니다. 결합 길이에 대한 PBE 기능의 과대평가는 이전에 간략하게 보고된 바와 같이 실험 결과와 비교하여 분산 보정으로 극복됩니다[1]. 카올리나이트의 구조에 대한 TM 도핑의 영향을 구별하기 위해 여기에서 먼저 격자 구조와 중심 양이온(Si 및 Al)과 산소 원자 사이의 최적화된 결합 거리인 Oa를 다시 살펴봅니다. , Ob 및 OH여관 .
표 1에서 볼 수 있듯이 카올리나이트의 경우 분산 보정된 PBE-D2 기능을 사용하여 최적화된 계산된 단위 셀 부피가 실험 값에 가깝고 PBE 기능(~3.4%)에 비해 상대 오차(~0.4%)가 현저히 낮아졌습니다. . 격자 벡터 a와 b의 경우 PBE-D2를 사용한 상대 오차(~0.4%)는 PBE(~1.1%)보다 훨씬 낮습니다. 그리고, PBE-D2의 분산 보정 하에서, 카올리나이트의 층간격(벡터 c)은 0.17 Å(~2%) 감소하였다. 특히, 분산 보정 후 격자 각도는 특히 α의 경우 실험 결과에 매우 가깝습니다. PBE-D2는 Si-Oa에 대해 거의 개선되지 않지만 카올리나이트의 결합 길이 분포에 관해서는 , Al–OH내부 , 및 Al-O 결합은 실험 결과와 비교하여 Al-OHinter Al-O 표면에서의 결합(표면 화학에 중요) 및 Si-Ob에 대한 약간의 개선 SiO 표면에서 결합. 특히 Al–OHinter의 경우 본드에서 PBE-D2의 분산 보정은 Al-O 표면의 최외곽층에서의 결합 환경을 정확하게 설명하는 것으로 보이며, 이는 위에 있는 다른 카올리나이트 층의 Si-O 표면으로부터의 분산력에 의해 크게 영향을 받습니다. 여기서 언급할 또 다른 요점은 실제로 약 1.95 및 2.00Å의 상당히 다른 결합 길이를 가진 두 개의 분할된 Al-O 결합(그림 1, 파란색 점선)이 있다는 것입니다. 이는 Al-O의 격자 왜곡을 보여줍니다. 팔면체는 Si-O 시트와 Al-O 시트 사이의 격자 불일치에서 비롯됩니다. 실험 결과와 비교하여 카올리나이트 구조 계산의 주요 오류로서 이러한 Al-O 결합은 평균 결합 길이가 유사한 PBE와 PBE-D2 모두에 의해 과대 평가되었습니다(표 1). PBE-D2는 약 1.96 및 2.04Å의 두 Al-O 결합을 제공하며 두 번째 결합은 0.04Å으로 과대평가됩니다(그림 2, 파란색 점선).
<그림>
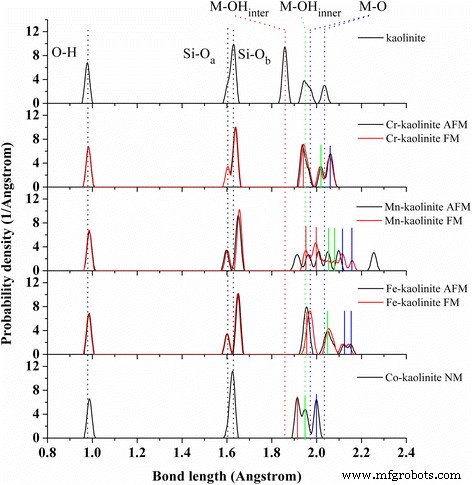
Cr–, Mn–, Fe– 및 Co–카올리나이트의 결합 분포. 다중 자기 상태는 각 TM-카올리나이트에 대해 제공됩니다. 다른 유형의 OH(검정 ), Si-Oa (검정 ), SiOb (검정 ), M–OH인터 (빨간색 ), M–OH내부 (녹색 ) 및 M–O(파란색 ) 카올리나이트의 결합은 점선으로 표시됩니다. . M–OH인터 (빨간색 ), M–OH내부 (녹색 ) 및 M–O(파란색 ) Cr-카올리나이트의 결합(AFM ), Mn-카올리나이트(FM ), Fe-카올리나이트(AFM ), 및 Co-kaolinite(NM )은 실선으로 표시됩니다.
전이 금속(Cr, Mn, Fe 및 Co) 도핑된 카올리나이트는 Al 원자를 Cr, Mn, Fe 또는 Co 원자로 대체하여 구성되었습니다. Al
3+
의 동등한 치환만 이온 TM
3+
+3 이외의 화학적 상태로 TM 이온을 동등하지 않게 치환하면 전하 균형을 위해 추가적인 공석이나 불순물이 발생하기 때문에 이온이 고려되었습니다. 구조적 관점에서, TM-카올리나이트의 PBE 및 PBE-D2 작용기는 카올리나이트에서 관찰된 것과 유사한 구조 차이를 제공합니다. PBE-D2 기능이 TM-카올리나이트에 대한 논의 후 카올리나이트의 두 기저 표면의 격자 벡터 및 결합 길이에 대해 더 잘 설명한다는 점을 고려하면 주로 PBE-D2 기능에 의해 얻은 결과에 의존했습니다. TM 도핑된 카올리나이트의 격자 매개변수, 결합 길이, 전하 및 스핀과 이들의 자기 상태는 표 1에 요약되어 있습니다. Cr-카올리나이트, Mn-카올리나이트 및 Fe-에 대한 AFM 및 FM 상태 간의 에너지 차이(TM 원자당) 카올리나이트는 각각 0.022, -0.006, 0.094eV입니다. Co-kaolinite 구조는 비자성 상태에서만 안정하므로 Co-kaolinite의 NM 구조만 표시됩니다.
TM-카올리나이트의 단위 셀 부피는 카올리나이트에 비해 확장되었으며, Mn-카올리나이트> Fe-카올리나이트> Cr-카올리나이트> 카올리나이트> Co-카올리나이트의 경향이 있습니다. 세포 확장은 주로 Al-O 결합에 비해 더 긴 M-O 결합으로 인해 발생하며, 이는 격자 벡터 a 및 b의 주요 확장으로 이어집니다. 한편, Si-Ob Si-O 시트의 결합은 동시에 늘어나며, 이에 따라 α 및 β의 결정 격자 각도가 왜곡됩니다. FM 상태의 Mn-카올리나이트의 셀 부피는 AFM 상태와 비교하여 1.4% 증가하는 반면 Cr-카올리나이트 및 Fe-카올리나이트의 셀 부피에 대한 자기 정렬의 영향은 거의 발견되지 않습니다. Cr, Mn, Fe 및 Co의 자기 모멘트는 TM 도핑된 Al2의 자기 모멘트에 가깝습니다. O3 [33], Mulliken 전하가 약간 높으므로 반응성이 더 강합니다.
TM-kaolinite의 결합 길이 분포는 각 도핑 요소에 대해 실선으로 표시된 TM-kaolinite의 다양한 유형의 Si-O 및 M-O 결합과 함께 그림 2에서 분석됩니다. 전반적으로 말하면, M–O와 Si–Ob의 결합 길이가 증가합니다. TM 도핑 후 M–OHinter에 대한 분할된 M–O 결합의 결합 분포가 재구성됩니다. (빨간색), M–OH내부 (녹색) 및 M–O (파란색) 결합. 특히, 분할된 Al-O 결합(파란색 점선)은 Cr 및 Co 도핑 후에 사라졌습니다. 또한, 결합 길이 분포는 Mn 원자의 자기 순서에 크게 의존하지만 Cr 및 Fe 원자의 경우 약간만 영향을 받습니다.
Cr
3+
에 대한 PDOS 결과 (d3), Mn
3+
(d4), Fe
3+
(d5) 및 공동
3+
(d6) 및 해당 전하 밀도 분포가 그림 2에 나와 있습니다. 3과 4. Jahn-Teller 정리에 따르면, 모든 퇴화 전자 시스템은 주변 결합 환경[47]의 영향을 받는 퇴화[46]를 제거하는 방식으로 자발적으로 왜곡됩니다. TM
3+
용 많은 수산기가 있는 카올리나이트의 팔면체 Al 부위에 도핑, TM
3+
의 5개 d-쉘 궤도 삼중항 t2g로 분할됩니다. 상태 및 이중선 eg 오 대칭 상태입니다. 삼중항 상태의 전자는 리간드 사이의 중간 영역에 국한되고 가장 가까운 O 상태와 더 혼성화됩니다. 이중선 상태의 것들은 리간드를 직접 가리키므로 t2g보다 에너지가 더 높습니다. 전자. 일반적으로 eg에 전자의 존재 오비탈은 팔면체 결합을 불안정하게 만드는 경향이 있으며, 채워진 오비탈 반대쪽의 결합을 늘리고 빈 오비탈 반대쪽의 결합을 단축하여 축퇴를 제거합니다. TM
3+
의 d-d 전환 (오) 종은 항상 점유된 t2g에서 나옵니다. 비어 있는 eg에 대한 궤도(dxy, dyz 및 dzx) 궤도(dx2-y2 또는 dz2 , 점유에 따라 다름). eg 사이의 궤도 분할 궤도 및 t2g Cr
3+
의 오비탈 (d
3
), Mn
3+
(d
4
), Fe
3+
(d
5
) 및 공동
3+
(d
6
) TM-kaolinite의 경우 Al2의 경우와 유사합니다. O3 및 TiO2 [33, 48, 49], 그러나 3차원 오비탈 사이의 분할 에너지는 자체 산화물보다 약간 더 크며(그림 3), 아마도 주변 히드록실기와의 혼성화 때문일 수 있습니다.
<사진>
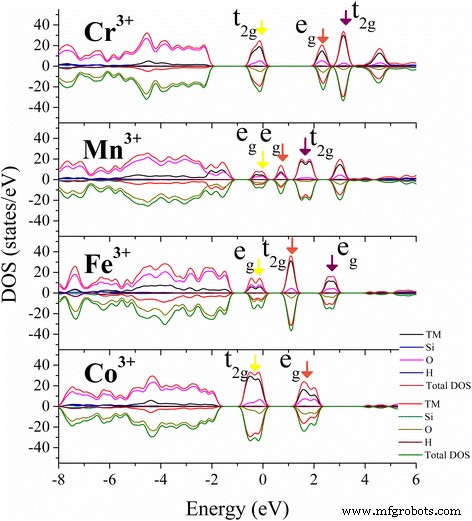
TM 도핑된 카올리나이트에 대한 가장 안정적인 상태의 총 상태 밀도(DOS) 및 원자 투영 상태 밀도(PDOS)가 제공됩니다. 가장 높은 점유 3D 오비탈(노란색 ) 및 첫 번째(갈색 ) 및 두 번째(보라색 ) 페르미 준위 주변의 가장 낮은 비어 있는 3d 궤도는 색 화살표로 표시됩니다.
<그림>
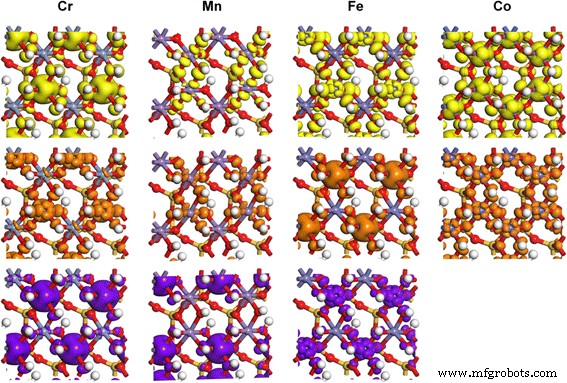
화살표가 가리키는 상태에 해당하는 TM-카올리나이트에서 TM 3d 궤도의 부분 전하 밀도 PDOS 결과에서. 등가곡면 수준은 0.02e/Å
3
입니다.
Mn-kaolinite의 FM과 AFM 상태 사이의 분열 에너지 차이는 작고 상태 밀도 분포는 스핀 방향이 다른 것을 제외하고는 유사합니다. 따라서 단순화를 위해 AFM 상태에 대한 결과만 표시됩니다. 고회전 Mn
3+
의 경우 (d
4
) AFM 상태의 Mn-kaolinite 이온, 두 eg 중 하나만 오비탈은 가전자대 최대값(VBM)에서 점유됩니다(그림 3, 노란색 화살표). dz2의 직업 에너지가 더 낮은 궤도는 z를 따라 두 리간드의 결합 전자에 강한 반발력을 줍니다. 축을 만들고 그 방향으로 M-O 결합을 늘립니다. 이 효과는 잘 알려진 Jahn-Teller 효과입니다. 전도대 최소값(CBM) 하단의 상태는 가장 낮은 비점유 dz2로 구성됩니다. 궤도(갈색 화살표) 및 더 높은 dx2y2 Mn
3+
의 오비탈(보라색 화살표) (d
4
). Cr
3+
의 경우 (d
3
), Fe
3+
(d
5
) 및 공동
3+
(d
6
) 도핑된 경우, 여기서 t2g 그리고 eg 궤도가 고르게 점유되고 Jahn-Teller 왜곡 효과의 영향이 작아 TM-kaolinite에서 M-O 결합의 약간의 편차만 발생했습니다(그림 2). TM 도핑에 의한 이러한 구조 및 전자 특성의 수정은 촉매 [50, 51], CO 포집 [52, 53], 약물 로딩 [54] 및 에너지 저장 [55,56,57] 분야에서 카올린의 적용을 향상시킬 수 있습니다. ]. 또한 몬모릴로나이트[50, 58], 펄라이트[55], 활석[59]과 같은 다른 광물에도 적용하여 전자적 특성을 변경할 수 있습니다.
결론
카올리나이트 나노클레이의 기하학적 구조와 전자구조에 대한 전이금속(Cr, Mn, Fe, Co) 도핑의 영향을 DFT 계산으로 조사하였다. 결정 부피, 격자 매개변수, 결합 길이, 전하 및 스핀, 가능한 자기 상태를 계산하고 연구합니다. Cr
3+
및 Fe
3+
도펀트는 AFM 상태, Mn
3+
에서 더 안정적입니다. FM 상태 선호 및 Co
3+
도펀트는 NM 상태를 선호합니다. 전이 금속 도핑은 격자 부피 팽창과 M-O 결합 분포의 일부 재구성을 유도합니다. 한편, TM 도펀트는 카올리나이트의 밴드 갭에서 더 큰 분할 에너지를 가진 일부 3차원 상태를 도입합니다.