

제조공정

산업 제조
직접 환원 과정의 이론적 측면
철광석의 직접환원법에서는 철광석이나 금속을 융해하지 않고 고체철광석으로부터 고체금속철(Fe)을 직접 얻는다. 직접 환원은 산화철의 환원을 허용하지만 다른 산화물(MnO, SiO2 등)은 환원할 수 없는 산소(O2) 전위에서 고체 상태의 환원으로 정의할 수 있습니다. 환원은 고체 상태이므로 환원된 철에서 이러한 원소가 용해될 가능성이 매우 적기 때문에(낮은 열역학적 활성에서) 철보다 더 안정적인 산화물은 본질적으로 환원되지 않은 채로 남아 있습니다. 철광석의 직접적인 환원은 상승하는 가스에 의해 용광로의 샤프트에서도 발생합니다.
철 - 산소 시스템
철-산소(Fe-O) 시스템은 아마도 가장 광범위하게 연구된 시스템 중 하나일 것입니다. 시스템의 열역학은 잘 이해되고 있으며 산화철과 관련된 기체 환원의 역학에 대한 많은 정보를 이용할 수 있습니다. 1 kg/sq cm의 총 압력에서 Fe-O 시스템에서 400℃와 1400℃ 사이에서 발생하는 열역학적으로 안정적인 고체상은 이진 도표(그림 1)에 나와 있습니다. 이 다이어그램은 Fe가 O2와 함께 세 가지 안정한 고체 화합물, 즉 (i) 적철광 - Fe2O3, (ii) 자철광 - Fe3O4 및 wustite - FexO를 형성한다는 것을 보여줍니다. 여기서 x는 1보다 약간 낮습니다. 비화학량론적 FeO 상( wustite)는 570℃ 이하에서 불안정하며 금속 Fe와 Fe3O4의 혼합물로 분해됩니다. 따라서 570 deg C 미만의 일정한 온도에서 위상 다이어그램을 가로질러 오른쪽에서 왼쪽으로 읽으면 상 순서는 Fe2O3 – Fe3O4 – Fe인 반면, 570 deg C 이상에서는 순서가 Fe2O3 – Fe3O4 – FeO – Fe입니다.
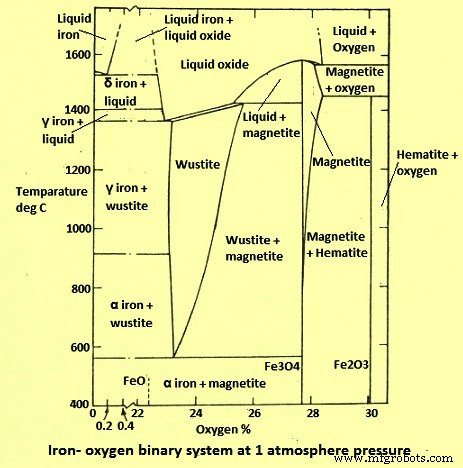
그림 1 Fe-O 바이너리 시스템 다이어그램
고체 알파 및 감마 철에서 O2의 무시할 수 있는 용해도는 O2의 0.01% 미만입니다. 따라서 O2 함량은 고체 Fe 변형의 전이 온도에 영향을 미치지 않으며 다이어그램에서 무시됩니다.
반응 평형을 고려할 때, Fe 산화물의 환원은 (i) 적철광(Fe2O3) -> 자철광(Fe3O4), (ii) 자철광(Fe3O4) -> 철(Fe), (iii) 자철광( Fe3O4) -> 황철석(FeO), (iv) 황철석(FeO) -> 철(Fe)
Wustite는 570℃보다 높은 온도에서만 안정합니다. 위 반응에 대한 열역학적 평형은 수소(H2)와 일산화탄소(CO)라는 두 가지 주요 기체 환원제로 잘 알려져 있습니다.
철 – 산소 – 탄소 시스템
CO 및 CO2(이산화탄소) 가스와 고체 탄소(C)가 혼합된 Fe 및 Fe 산화물의 평형은 그림 2에 나와 있습니다.
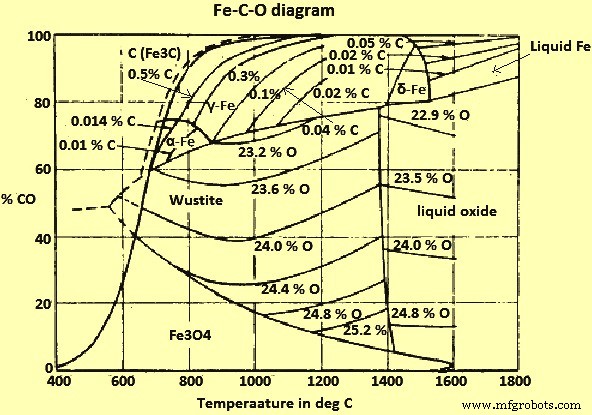
그림 2 Fe-CO-O 시스템 다이어그램
그림 2로부터 710℃ 이상의 온도와 1kg/sq cm의 총 압력에서 모든 Fe 산화물은 C와 평형을 이루는 CO/CO2 가스 혼합물에 의해 환원될 수 있으며, 따라서 C 자체에 의해 감소됩니다. 더 낮은 온도에서는 C로 과포화되어 부두아르 평형에 따라 C 증착을 향해 반응하는 혼합물만이 Wustite에 환원 작용을 합니다.
철 – 수소 – 산소 시스템
기체 H2와 H2O(증기)가 혼합된 Fe 및 Fe 산화물의 평형 다이어그램은 (그림 3)에 나와 있습니다.
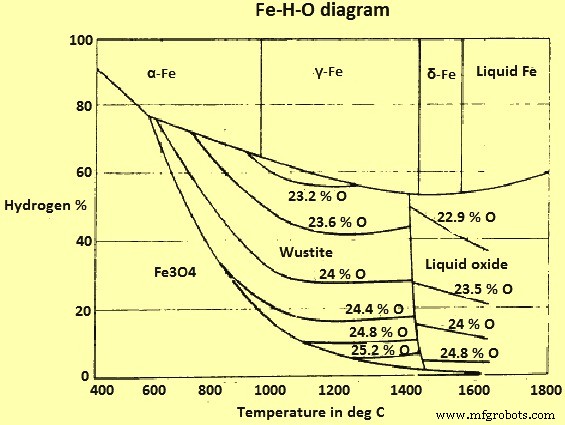
그림 3 Fe-H-O 시스템 다이어그램
이 시스템과 Fe-O-C 시스템의 주요 차이점은 '그을음 라인' 또는 해당 현상이 없다는 것입니다. 따라서 이론적으로 적철광(및 자철광)을 모든 온도에서 H2와 함께 Fe로 환원하는 것이 가능합니다.
CO와 H2에 의한 환원 비교
Fe-C-O 및 Fe-H-O 시스템에 대한 연구(그림 4)에서 815℃ 이상에서 H2는 CO보다 더 효율적인 환원제인 것으로 보입니다(즉, 평형 H2/H2O 비율은 상응하는 CO보다 낮습니다) /CO2 비율), 낮은 온도에서는 반대입니다. 그러나 이러한 평형은 평형에 접근함에 따라 감소 속도가 매우 느려지기 때문에 산업용 용광로에서는 거의 달성되지 않습니다. 조건이 평형에서 분명히 벗어날 때 H2 및 CO를 사용한 환원에 대한 각각의 반응 속도는 평형 고려에서 일반적으로 예상되는 것과 반대 순서입니다. 따라서 H2는 실제로 815°C보다 낮은 온도에서 작동하도록 설계된 비평형 공정에 대해 더 효율적인 환원제이고 CO는 더 높은 온도에서 더 효율적입니다.
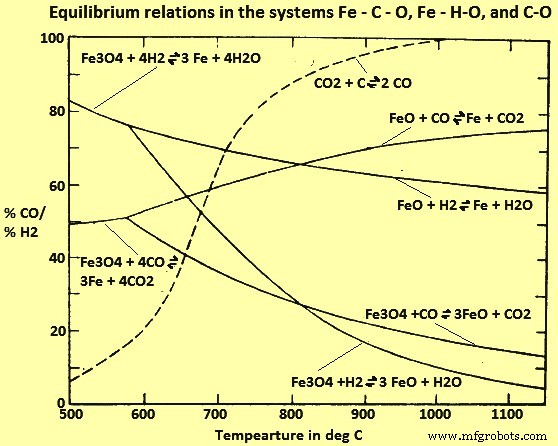
그림 4 Fe-C-O, Fe-H-O 및 C-O 시스템의 평형 관계
다른 온도에서 CO와 H2의 가스 조성 혼합물의 영향에 대한 연구는 환원 가스 혼합물의 H2 함량이 증가함에 따라 반응 속도가 증가하는 것으로 나타났습니다. 이 관계는 현저하게 비선형적인 것으로 밝혀졌습니다.
Fe 산화물을 H2와 함께 금속 Fe로 환원하는 것은 흡열이며 필요한 온도를 유지하기 위해 외부 열원이 필요합니다. CO와의 해당 반응은 발열 반응이며 적절하게 제어된 조건에서 반응은 열적으로 자립적입니다. 사실, 충전물의 과열을 피하기 위해 H2 또는 기타 열 흡수 가스로 CO를 희석해야 할 수도 있습니다. 일부 공정은 CO-H2 열 균형을 이용하고 이러한 가스의 혼합물을 사용하여 주변 온도에서 최대 반응 온도인 약 1100℃까지 광석을 가열하는 동안 얻어지는 환원량을 증가시키도록 설계되었습니다.
그림 4는 기체 환원이 경제적으로 가능한 범위 내의 모든 온도에 대해 평형 기체 혼합물이 최소 60%의 CO 및/또는 H2를 함유함을 보여줍니다. 평형에 도달하지 못하면 이러한 가스의 미반응 농도는 훨씬 더 높아지고 대부분은 환원로를 통해 그대로 통과합니다. 공정이 경제적인 경우에는 Wustite를 금속 Fe로 환원, 고급 Fe 산화물을 Wustite로 환원 및/또는 가스 혼합물 및 기체 반응 생성물의 제거.
기체-고체 반응 및 고체-고체 반응
기체-고체 반응은 기술에서 중요한 역할을 하며 광석에서 금속 추출(Fe 산화물 환원 등)을 포함하여 매우 광범위한 분야를 포함합니다. 모든 기체-고체 반응 시스템의 공통된 특징은 전체 공정이 여러 중간 단계를 포함할 수 있다는 것입니다. 일반적으로, 이러한 중간 단계는 (i) 대부분의 기상에서 반응하는 고체 입자의 외부 표면으로 반응물 및 생성물의 기체 확산(질량 전달), (ii) 기체 반응물 또는 기체 생성물의 기공을 통한 확산을 포함합니다. 고체 반응 생성물 또는 부분적으로 반응된 고체의 기공을 통한 (iii) 고체 표면에서의 기체 반응물의 흡착 및 반응 생성물의 탈착, (iv) 흡착된 기체와 고체 사이의 실제 화학 반응.
기체-고체 반응 영역에는 기체-고체 반응이 수행되는 노의 성능과 반응 진행에 영향을 줄 수 있는 몇 가지 다른 현상이 있습니다. 이러한 다른 현상에는 열 전달, 반응에 수반되는 고체 구조의 변화(예:소결), 기체-고체 반응이 일어나는 용광로를 통한 기체 및 고체의 흐름이 포함됩니다. 감소율은 사용된 프로세스에 따라 이러한 요소에 의해 제어됩니다.
고체 사이의 반응은 두 가지 주요 그룹으로 나눌 수 있습니다. 즉 (i) 진정한 고체 – 서로 접촉하는 두 입자 사이의 고체 상태에서 일어나는 고체 반응 또는 고체 상태의 입자 이동(예:형성 Fe 산화물과 C) 사이의 반응을 통한 탄화철의 반응 및 (ii) 기체 중간체를 통해 일어나는 고체 반응물 사이의 반응(예:1kg/sq cm 압력에서 탄소로 Fe 산화물 환원)
고체 C를 사용한 Fe 산화물의 환원은 매우 낮은 절대 압력에서 수행된다면 진정한 고체-고체 반응이 될 수도 있습니다. 0.0005mm Hg(수은)의 진공 하에서 미세 분말 흑연(C)과 적철광 광석의 혼합물의 반응에 의해 수행된 연구 중 하나에서 최대 900℃의 온도에서 반응이 매우 일어난다는 것이 발견되었습니다. 천천히, 그리고 18시간 안에 Fe3O4와 FeO만 형성되었지만 Fe는 형성되지 않았습니다. 테스트 동안 더 높은 온도에서만 눈에 띄는 변형이 관찰되었습니다. 연구 중에 반응 속도는 산화물 상 내에서 Fe 이온의 확산에 의해 결정된다는 결론을 내렸습니다. 다른 조사에서 나온 추론은 C가 Fe 산화물에서 확산되고 아마도 역사적 관심의 대상일 뿐이라는 것입니다. 그러나 이러한 연구는 분말 혼합물 위의 기체 압력이 증가할 때 반응 속도가 뚜렷하게 증가한다는 것을 보여주었습니다. N2(질소)의 흐름이 C와 Fe 산화물의 혼합물을 통과하는 유사한 유형의 테스트에서 N2의 흐름이 증가함에 따라 반응 속도의 현저한 감소가 관찰되었습니다. CO 또는 H2에서 유사한 분말형 Fe 산화물의 환원 속도와 함께 진공 또는 N2 하에서 수행된 이러한 모든 조사는 C와 광석의 직접적인 고체 상태 반응(때때로 실제 메커니즘으로 간주되는 진정한 직접환원의 정도)는 공업로에서 환원공정의 진행에 있어 중요하지 않다.
환원철의 기공 구조
여러 천연 광석에 대한 환원성 테스트는 철광석 입자의 다공성이 환원성을 제어하는 가장 중요한 요소 중 하나임을 보여주었습니다. 90% 환원에 필요한 시간의 역수로 표현되는 환원성은 다공성에 따라 직접적으로 변합니다. 상대 환원성은 "상대 환원성 =(다공도 x 0.75) + 8.0" 방정식에 의해 주어진 것처럼 다공성이 증가함에 따라 증가합니다.
Fe 산화물의 환원은 항상 다공성 반응 생성물을 생성합니다. 산화물의 성질과 환원 조건은 환원철의 기공 구조에 영향을 미친다. 이는 입자의 표면에서 안쪽으로 환원이 진행되기 때문이다. 원래의 우스타이트 표면에 의해 정의된 체적 점유 공간이 감소됩니다. 이것은 다공성을 개발해야만 달성할 수 있습니다. 이 다공성에 대한 주사 전자 현미경 연구는 일반적으로 H2 환원이 CO 환원에 의해 얻어진 것보다 더 미세한 기공 구조를 제공한다는 것을 보여주었습니다. 또한, 주사형 전자현미경 사진에서 H2 환원 온도가 600℃에서 1200℃로 점진적으로 증가함에 따라 기공 구조가 조대해지는 것이 명백해진다.
Fe 산화물의 초기 세공 표면적은 기체 환원에 의해 형성된 환원철의 세공 표면적에 영향을 미친다. Fe 산화물의 초기 세공 표면적의 감소는 환원철의 세공 표면적을 감소시킨다. BET(Brunauer-Emmett-Teller) 기술로 측정한 H2의 적철광 광석에서 환원된 철의 공극 표면적은 환원 온도가 증가함에 따라 감소하는 것으로 나타났습니다.
환원 온도와 평균 임계 기공 크기 및 최소 기공 반경 사이의 관계는 기공 크기 분포로부터 얻어졌다. 기공 크기는 환원 온도가 900℃까지 증가함에 따라 천천히 증가하는 것으로 밝혀졌지만 온도가 더 증가함에 따라 급격히 증가했습니다. 이러한 결과는 900℃ 이상의 환원 온도에서 기공 구조의 뚜렷한 조대화를 보여주는 주사 전자 현미경에 의한 파단 표면 관찰에 따른 것입니다.
환원 적철광 광석의 공극 표면적은 환원 온도와 가스 조성의 영향도 받습니다. CO/CO2 가스 혼합물에서 환원된 적철광에서 얻은 공극 표면적은 H2/H2O 가스 혼합물에서 환원된 것의 약 2/3입니다. 이는 현미경으로 관찰한 CO/CO2 환원철의 거친 기공 구조와 일치합니다.
환원철 기공의 가스 확산이 측정되었습니다. 다공성 매질의 확산 플럭스는 (i) 압력과 무관하고 T(온도)의 1/2승에 비례하는 크누센(Knudsen) 확산과 (ii) 압력에 반비례하고 T에 비례하는 분자 확산이라는 두 가지 확산 과정을 통해 발생합니다. 전력 3/2.
제한적인 이상적인 구조는 45도 각도로 서로 연결되고 교차하는 균일한 크기의 기공을 갖는 것으로 가정됩니다.
효과적인 확산율은 온도와 압력에 따라 주어진 다공성 매질에 따라 달라지며 서로 다른 이원 기체 쌍에 따라 다릅니다. 환원 온도가 낮아질수록 기공 구조가 미세해집니다.
감소 모드
천연 철광석 입자 또는 소결 적철광 펠릿의 환원은 제품 층을 형성합니다. 이 잘 알려진 현상은 많은 연구의 주제였습니다. H2에 의한 소결된 적철광 펠릿의 환원에 대한 최근 연구 중 하나에서 부분 환원된 적철광 펠릿의 연마된 부분에 층 형성의 전형적인 예가 있음이 주목되었습니다. 레이어 사이의 비교적 부드러운 인터페이스는 일반적으로 낮은 배율에서 나타나지만 이러한 모양은 오해의 소지가 있습니다.
이것은 가스 확산이 Wustite 층에서 충분하여 전진하는 Fe/FeO 계면에 앞서 약간의 내부 환원을 제공한다는 것을 나타냅니다. 내부 환원 영역은 (i) 온도가 낮아지고, (ii) 다공성이 증가하고, (iii) 입자 크기가 작아질수록 확장됩니다.
주어진 감소 비율을 달성하는 데 필요한 시간에 대한 입자 크기의 영향은 감소 모드 및 따라서 속도 제어 프로세스 유형에 따라 다릅니다. 기체 환원에 의한 다공성 Fe 산화물의 환원 모드를 고려하면 (i) 균일한 내부 환원, (ii) 혼합 제어 제한, (iii) 다공성 Fe 층에서의 확산이라는 세 가지 제한 속도 제어 프로세스가 나타납니다. 환원이 이들 중 어느 하나에 의해 단독으로 제어되는 경우, 환원 시간은 세 가지 방식 중 하나로 입자(구형) 지름과 관련됩니다. 즉 (i) 시간이 지름과 무관한 균일한 내부 환원, (ii) 혼합 제어 제한 및 (iii) 다공성 철의 확산.
속도 조절 과정은 (i) 균일한 내부 환원이 있어 작은 입자 크기가 필요하거나 (ii) 입자 크기가 크기 때문에 철층의 기공에서 가스 확산에 의한 궁극적인 속도 조절이 우세할 때만 비교적 간단합니다. 크다. 또한 온도, 가스 조성, 입자 크기 및 산화물 유형에 따라 환원이 진행됨에 따라 하나의 제한 속도 제어 공정에서 다른 제한 속도 제어 공정으로의 전환이 있을 수 있음을 이해해야 합니다. 산화철의 환원은 또한 일부 설명할 수 없고 비정상적인 행동을 보일 수 있습니다.
다공성 철광석 입자 감소율
광석의 기공률과 기공 구조는 내부 환원의 정도와 균일성에 현저한 영향을 미칩니다. 연구 중 하나에서 90% CO와 10% CO2 혼합물에 대한 적철광 광석의 환원율에 대한 입자 크기의 영향과 1000℃에서 H2에 대한 영향은 입자 크기가 증가함에 따라 내부 환원이 제한된다는 것을 보여주었습니다. 따라서 입자 크기가 증가함에 따라 전체 감소율이 감소합니다.
다공성 적철광 입자의 환원 초기 단계에서 FeO로의 급속한 전환 후 FeO에서 Fe로의 내부 환원이 뒤따른다. Fe 산화물의 기공에서 거의 완벽한 기체 확산의 제한적인 경우, 내부 환원이 우세하고 속도는 주로 기공 벽에서의 기체-고체 반응에 의해 제어됩니다. 몇 개의 원자 두께인 Fe 층이 FeO의 기공 벽을 덮고 있다고 가정합니다. 환원율은 기공벽의 Fe 층의 코팅을 통한 O2의 급속한 확산과 이 매우 얇은 Fe 층 표면의 O2와 H2 또는 CO의 화학 반응에 의해 공동으로 제어되는 것으로 추정됩니다.
입자 크기의 효과는 입자 크기가 감소함에 따라 감소 속도가 증가함을 보여줍니다. 일반적인 현미경 사진은 환원 모드가 입자 내에서 하나의 입자에서 다른 입자로 변한다는 것을 나타냅니다. 이것은 산화물 입자의 다공성의 국부적 차이 때문입니다. 기공 크기의 변화와 더 큰 기공에서 더 빠른 기체 확산으로 인해 대부분의 반응은 더 큰 기공의 벽에서 발생합니다. 즉, 전체 기공 표면적의 일부만이 반응에 사용될 것으로 예상됩니다. 다양한 유형의 적철광 광석 입자로 달성된 800℃에서 H2 환원 속도는 형성된 Fe(또는 FeO)의 기공 표면적이 증가함에 따라 비선형적으로 증가합니다. 이러한 결과는 기공 표면적이 클수록 반응에 사용된 총 기공 벽의 비율이 더 작다는 사실을 입증합니다.
H2 – CO 가스 혼합물의 내부 환원 속도는 일반적으로 H2와 CO에 대한 두 가지 개별 환원 속도의 합입니다. 가스 평형이 느립니다.
철광석(덩어리 또는 펠릿) 감소율
덩어리 광석 또는 광석 펠릿의 환원 속도는 충전층의 환원 가스 흐름에서 복잡한 성질을 가지고 있으며, 복잡성은 전체 환원 속도가 열 및 질량과 같은 일련의 여러 반응 공정에 의해 제어되기 때문입니다 기체막 경계층을 통한 전달, 기체-고체 반응 및 다공성 제품 층에서의 기체 확산. 컴퓨터 계산에 의해 촉진된 수학적 분석을 통해 다양한 환원 모드에 대한 큰 산화물 입자의 환원 속도를 설명하기 위해 수많은 방정식이 도출되었습니다.
단일 펠릿 또는 철광석 입자를 사용한 여러 실험에서 열 전달은 비교적 빠르며 충분히 높은 속도의 기체 흐름에서 기체 필름 물질 전달 저항은 무시할 만큼 작습니다. 따라서 환원 속도에 영향을 미치는 두 가지 주요 반응 단계, 즉 (i) 가스 산화물 반응, (ii) 다공성 산화물 및 다공성 생성물 층에서의 가스 확산이 있습니다. 이러한 속도 프로세스의 상대적 효과는 입자 크기, 가스 구성, 온도 및 환원 모드에 따라 달라지며 환원의 진행에 따라 변경됩니다.
다공성 Fe 층의 가스 확산
연구 중 하나에서 Fe 층의 기공에서 가스 확산의 영향을 입증하기 위해 단방향 환원 실험이 수행되었습니다. 긴 실린더 샘플은 덩어리 적철광 광석의 큰 조각으로 준비되었으며 꼭 맞는 니켈 튜브 안에 포장되었습니다. 필요한 시간 동안 H2를 감소시킨 후, 샘플을 축방향으로 분할하고 연마하고, Fe 층의 두께를 결정하였다. 실험 결과 환원된 Fe 층의 두께가 1mm 정도일 때 포물선율 법칙에 따라 추가 환원이 진행되며 이는 기공확산 조절 결과와 유사하다. 이러한 테스트는 다공성 Fe 층의 두께가 증가함에 따라 환원 속도가 결국 Fe 층의 기공 내 기체 확산에 의해 제어된다는 것을 입증했습니다.
Fe 층의 주요 전진 전면에 선행하는 부분적인 내부 환원은 환원된 층에 일부 FeO의 포획으로 이어질 수 있다. 이 상황은 환원의 마지막 단계에서 느린 O2 제거로 이어질 수 있습니다.
환원 온도가 감소함에 따라 기공 구조는 훨씬 더 미세해지며, 아마도 연결된 모세관에 많은 좁은 채널과 병목 현상이 있을 때 Knudsen 확산이 우세할 때 유효 분자 확산/유효 평균 Knudsen 확산 비율의 값이 낮습니다. 환원 온도가 증가함에 따라 기공 구조가 조대화되어 기공을 통한 기체의 통과가 용이해짐에 따라 비율이 높아집니다.
H2-CO-CO2 혼합물(CO/CO2 비율 포함)에 의해 900℃에서 환원된 소결된 적철광 광석 펠릿 및 마그네타이트 광석 펠릿의 50%, 75%, 90% 및 95% 감소를 달성하는 시간에 대한 가스 조성의 영향 그을음 침착을 억제하기 위해 9와 같음)은 H2가 CO로 대체됨에 따라 주어진 O2 제거 비율을 달성하기 위한 등온 환원 시간이 약 50% CO까지 점진적으로 증가하고 CO를 추가로 추가하면 감소의 시간. 100%(CO/CO2 비율 9)에 대한 환원 시간은 동일한 온도에서 H2보다 약 10배 더 깁니다. 기체의 운동 이론에서 파생된 H2-H2O 또는 CO-CO2와 같은 이원 시스템의 분자 기체 확산성은 시스템에 대해 불변이며 본질적으로 기체 조성과 무관합니다. 그러나 삼원 및 다중 구성 요소 시스템에서 각 종은 다른 확산성을 가지며 가스 구성에 따라 다릅니다. 또한, 확산 플럭스에 대한 속도 방정식은 복잡합니다.
H2-CO 혼합물에서 적철광 광석 펠릿의 환원 거동은 H2와 CO에서 관찰된 것과 유사한 패턴을 나타냈으며, 이는 약 50% O2 제거를 초과하는 환원율은 Fe 기공의 가스 확산에 의해 제어됩니다. 레이어.
초기 비율의 혼합 통제 제한
환원의 초기 단계에서 환원 속도는 (i) Fe2O의 기공에서의 기체 확산(FeO의 고체 상태 확산은 무시할 수 있음) 및 (ii) FeO의 기공 벽에서의 반응에 의해 공동으로 제어됩니다. . 이것은 얇은 다공성 Fe 층과 그 내부의 빠른 가스 확산을 의미합니다. FeO의 다공성과 그 안의 기체 확산도에 따라 공칭 Fe/FeO 경계면보다 앞서 부분적인 내부 환원이 있습니다. 다공성 FeO와 H2의 반응은 일반적으로 공칭 Fe/FeO 계면에 가까운 기공 입구에 국한됩니다.
일부 내부 축소
가스 구성, 온도, 펠릿 크기 및 총 가스 압력에 따라 제한 속도 법칙의 틀 내에서 감소의 일정 기간 동안 혼합 속도 제어가 있습니다. 속도 방정식은 일반적으로 펠릿의 기체 환원이 펠릿의 입자 간 기공을 통한 기체의 느린 역류 확산에 의해 공동으로 제어되고 기체와 Fe 산화물의 느린 화학 반응에 의해 공동으로 제어된다는 가정에 기반합니다. 입자의 Fe 산화물-/Fe 계면.
수성 가스 이동 반응
수성 가스 이동 반응은 산화철 환원에서 환원제로 개질된 탄화수소를 사용하는 직접 환원 공정에서 중요한 역할을 합니다. 일반적으로 CO 및 H2에 의한 철광석 환원율의 차이와 CO/CO2 혼합물에 함유된 소량의 H2가 환원율에 미치는 현저한 효과로부터 H2가 실제 환원 성분이라는 데 일반적으로 동의합니다. 이러한 가스 혼합물에서. CO는 주로 생성된 증기(H2O)를 다시 H2로 환원시키는 역할을 하는 것으로 간주됩니다. 반응은 (i) H2 + FeO =H2O + Fe, (ii) H2O + CO =H2 + CO2입니다.
이 반응의 두 번째 하위 프로세스는 수성 기체 전환 반응으로 알려져 있습니다. 이 과정에 촉매가 필요하다는 것은 잘 알려져 있습니다. 철광석 환원에서는 모든 생성물(Fe3O4, FeO, Fe)이 가능한 촉매로 고려됩니다. 이들 중에서 특히 활성인 것은 고체 Fe이다. 따라서 H2를 포함하는 CO/CO2 혼합물에서 철광석의 환원 과정은 금속 Fe가 존재할 때 반응 순서로 이해해야 합니다. 하위 반응 (i) 적절한 환원은 Fe 산화물 표면에서 발생하는 반면 하위 반응 (ii), 수성 가스 반응에 의한 H2 재생은 Fe 표면에서 발생합니다.
두 하위 반응의 공간적 분리는 반응 참가자 중 하나에 의해 기체 확산 또는 표면 확산으로 발생하는 수송 과정에 의한 연결이 필요합니다. 최적의 조건은 3상 경계 Fe/Fe 산화물/가스에서 발생합니다.
축소 중 부기
철광석이나 펠릿의 겉보기 부피는 일반적으로 환원 중에 증가합니다. 이것을 붓기라고 합니다. 볼 수 있는 팽윤 거동은 크게 세 가지입니다. 이것은 (i) 정상적인 팽창, (ii) Fe2O가 Fe로 전환되면서 급격한 부피 팽창이 있는 치명적인 팽창으로 알려져 있으며, Fe는 섬유상 Fe의 위스커 와이어로 알려진 필라멘트 성장의 형태로 나타납니다. (iii) 소량의 알칼리를 포함하는 철이 풍부한 재료의 전형적인 거동인 파열 팽창. 이러한 후자의 행동은 Fe가 반응 생성물로 나타나기 전에 팽창의 대부분이 일어난다는 점에서 (심각한 정도는 아니지만) 치명적인 팽창과 다릅니다.
덩어리광석이나 소결석은 비정상적으로 팽창하거나 파멸적으로 팽창하는 것으로 알려져 있지 않은 반면, 특정 유형의 펠릿은 팽창하며, 비정상적으로 팽창된 펠릿은 부드럽고 해면질이며 분해되는 경향이 있기 때문에 부담의 투과성을 감소시켜 작업상의 문제를 야기한다고 말할 수 있습니다. .
문헌에 보고된 다양한 Fe 산화물 및 Fe의 특정 부피는 Fe 그램당 Fe2O3 0.272cc(실온에서), Fe 그램당 Fe3O4 0.270cc, Fe 그램당 FeO 0.231cc(23.5% O2)입니다. , 그리고 Fe 그램당 Fe 0.128cc. 따라서 각 감소 단계에서 볼륨이 감소할 것으로 예상됩니다. 그러나 Fe 광석의 팽윤의 주요 원인은 육각형 적철광 광석이 입방정 마그네타이트 광석으로 변태되어 결과적으로 격자 교란이 발생하기 때문입니다. 격자 교란은 기공 형성을 유발하여 적철광에서 자철광으로 변태하는 동안 Fe 광석의 겉보기 부피가 상당히 증가합니다.
일반적으로 CO가 풍부한 가스가 감소하는 동안 팽윤은 H2가 풍부한 가스보다 훨씬 더 큽니다. 이 거동의 이유는 CO 함유 가스 혼합물에서 C 증착 동안 금속 더스팅이 발생하기 때문입니다. 그러나 C 증착이 없을 때 CO-CO2 가스 혼합물의 환원 중에 발생할 수 있는 팽창을 설명하기는 어렵습니다. 축소에 따른 부기나 수축의 원인과 효과는 아직 해결되지 않았습니다.
일반적으로 광석 펠릿에는 두 가지 유형의 불순물이 있습니다. (i) 팽윤을 억제하는 불순물, (ii) 팽윤 개선 효과가 있는 불순물이다. 첫 번째 예는 실리카(SiO2)이고 두 번째 예는 알칼리(K2O, Na2O)입니다. 최대 5%의 SiO2를 포함하는 시약 등급 Fe2O3 펠릿은 CO – CO2 가스 혼합물에서 환원될 때 팽창하지 않으며 강도를 유지하고 치명적인 팽창을 방지하기 위해 산성 펠릿에 일정량의 SiO2가 필요합니다. 두 번째 경우, 0.1% ~ 1% 범위의 알칼리 Na2CO3 또는 K2CO3를 소량 첨가하면 일반 광석 펠릿의 H2 또는 CO에 치명적인 팽창이 발생할 수 있음을 알 수 있습니다. 알칼리의 영향은 펠릿의 염기도(CaO/SiO2) 비율이 증가할수록 더욱 두드러집니다. 미세한 산성 맥석을 첨가하여 안정적인 알칼리 규산염을 형성함으로써 부작용을 예방할 수 있습니다.
광석 펠릿의 불순물 영향(예:석회 함량)에 대해 모순되는 관찰이 있습니다. 적철광 광석 펠릿에 소량의 CaO(0.1% 미만)를 첨가하면 환원 중에 상당한 팽윤이 발생하며 이는 CaO가 치명적인 팽윤의 원인임을 시사합니다. 한편, 적철광 광석 펠릿에 약 1% CaO를 첨가하면 환원 중 팽윤을 억제하는 것으로 나타났습니다. 팽윤에 대한 CaO의 관찰된 효과의 이러한 변화는 알칼리와 같은 철광석의 다른 불순물의 존재 또는 부재로 인한 것일 수 있습니다.
C에 의한 적철광 광석 환원
적철광 광석과 C 사이의 반응은 금속화된 광석 펠릿의 제조에서 근본적으로 중요합니다. 새로운 관심의 대부분은 DRI(직접환원철) 생산에서 환원제로 고체 C를 사용하는 회전식 가마 공정의 개발로 자극을 받았습니다. C에 의한 Fe 산화물의 환원은 진정한 고체-고체 반응이 지배적인 메커니즘인 매우 높은 진공 상태를 제외하고 기체 중간체 CO 및 CO2를 통해 발생한다는 것이 일반적으로 받아들여지고 있습니다.
C에 의한 적철광의 환원 동안 일어나는 기체 중간체를 통한 반응 메커니즘은 (i) C(s) + 0.5 O2 =CO(g), (ii) FexOy(s) + CO(g) =FexO 반응을 통해 이루어집니다. (y-1) (s) + CO2(g) 및 (iii) CO2(g) + C(s) =2CO(g).
CO의 초기 형성은 전체 반응 속도에서 중요한 단계입니다. 갇힌 공기의 O2는 Fe 산화물의 해리에 의해 방출된 O2 가스와 함께 C와 반응하여 CO를 생성합니다(첫 번째 반응). 또한 일부 CO는 C와 Fe 산화물 입자 사이의 접촉 지점에서 발생하는 진정한 직접 환원에 의해 형성될 수도 있습니다. 이렇게 생성된 CO 가스는 적철광 광석 입자와 쉽게 반응합니다(2차 반응). Boudouard 또는 CO2 가스와 C 입자 사이의 용액 손실 반응은 CO 가스를 재생성(3차 반응)하여 샘플의 기공 내에 포함된 기상의 환원 전위를 회복시키는 경향이 있습니다. CO2에서 특정 유형의 C의 산화는 특정 금속 및 금속 화합물의 존재 하에서 촉매됩니다. 공정의 속도 향상은 Li2O(산화리튬)의 첨가로 관찰되었으며 억제 효과는 FeS(황화제1철)의 첨가로 보고되었습니다. 금속 Fe는 흑연(C)의 가스화에 좋은 촉매인 것으로 밝혀졌습니다. 혼합물에서 예측할 수 없는 촉매 반응 때문에 전체 반응 속도를 설명하기 위해 수학적 모델링을 통해 유도된 방정식은 값이 제한적이며 반응이 촉매되지 않는 시스템에만 적용할 수 있습니다.
적당히 높은 온도(예:1000℃)에서 Fe 산화물 반응의 속도(570℃보다 높은 온도에서 Fe2O3, Fe3O4, FeO, Fe의 순서)는 Boudouard 반응의 속도보다 훨씬 더 큽니다. 즉, 전체 공정은 Boudouard 반응에 따른 CO 가스의 가용성에 의해 제한됩니다. 따라서 정상 상태에서 이 기상 조성은 FexOy/FexO(y-1)에 대한 평형 기상 조성과 밀접하게 일치합니다.
탄화수소를 사용한 산화철 환원
탄화수소는 DRI 생산을 위한 환원제로 두 가지 방식으로 사용될 수 있습니다. These are (i) direct use of hydro-carbons or a mixture of gas containing hydro-carbons, and (ii) use of the reformed hydrocarbon products (CO, H2), by reforming within the reduction reactor (it has been found that auto-catalytic reforming of some hydro-carbons within the reducing furnace provided an access of macro and micro porosity which leads to more extensive reduction and also which leads to the deletion of the capital cost of gas reformer and processing.
There are a few studies using directly hydrocarbons or a mixture of gas containing hydrocarbons as reductant for direct reduction of iron ores. Two important points emerge from these studies. The first is that the rate of reduction with hydrocarbons is slow and the production of a high quality of DRI is troublesome and uneconomical. The second point is that these studies have been done under isothermal conditions in a thermo-gravimeter with single particle or powder compact, thus the results are of only theoretical value.
Theoretical importance of investigations with hydrocarbons – The kinetics of ferric oxide reduction by pure methane (CH4) has been studied in the three temperature ranges of (i) low temperature (500 deg C to 600 deg C), (ii) medium temperature (650 deg C to 750 deg C) and (iii) high temperature (800 deg C to 950 deg C). At the low temperature, the reduction proceeds only from Fe2O3 to Fe3O4. A prolonged holding of the sample in a stream of CH4 has not led to any process extension beyond this stage. The rate became appreciable at 650 deg C. In special experiments after the Fe3O4 composition has been reached, the sample has been reduced further by H2 and CH4. It has been shown that CH4 reduction in the low temperature range beyond the Fe3O4 stage occurs only if a sufficient quantity of metallic Fe has been built up. In this case the reducing agent has not been CH4, but its decomposition product, H2. C formed by CH4 decomposition takes almost no part in the reduction and gets accumulated in the sample.
In the medium temperature range the conversion of Fe3O4 to FeO takes place but at low rates. A sharp rise in reduction rate is observed on going from 750 deg C to 800 deg C. The process becomes very sensitive to temperature changes beyond 800 deg C, and accelerated considerably in the high temperature range, when metallic Fe appeared in the sample. The appearance of metallic Fe at the FeO to Fe stage, at comparatively high temperatures indicates a decisive role of metallic Fe as a catalyst for reforming CH4 by the reduction products (CO2, and H2O). In the absence of a catalyst, the decomposition of CH4 and its reforming by the reduction products (CO2, H2O) do not occur to any substantial extent and no C accumulation in the sample has been observed. When the Fe catalyst is present, CH4 dissociation into the elements takes place only at very late stages of reduction, when there is insufficient CO2 and water vapour to convert all the CH4 diffused into the sample. C build-up in the sample starts from that stage.
In the 2-stage production of DRI with CH4, it has been found that the complete decomposition of CH4 in the presence of the Fe bearing material occurs at temperatures of 850 deg C to 900 deg C, which is 400 deg C to 450 deg C lower than on an inert surface (e.g. fire clay), while the reaction rate, conversely, has been 10 times higher. The products of the first stage are a sooty Fe containing 30 % to 50 % C and technically pure H2.
In the second stage, the product of the first stage (sooty Fe with highly dispersed C in the pores of DRI and on the surface of the Fe particles) has been used as an active reducing agent and mixed with mill scale or concentrate. The mixture has been reduced in the temperature range 1050 deg C to 1100 deg C with a make-up reducing agent of H2 reformed natural gas. The results of industrial trials has shown that the use of sooty Fe instead of soot, petroleum coke and the other known carbonaceous reducing agents considerably intensified the Fe-oxide reduction process. As is well known, the direct reduction of Fe oxides with C is directly related to the rate of reaction between the C and CO2. The sooty Fe can have intensified the rate of Boudouard reaction.
The isothermal reduction of hematite ore pellets (with 10 % to 15 % porosity) in a thermo-balance with a mixture of CH4-H2 (containing 4.5 % CH4) within the temperature range 700 deg C to 1000 deg C has shown that the reduction is chemical – controlled initially and diffusion – controlled in the later stages. It has been shown that reduction in pure H2 is faster than in the CH4- H2 mixture. This difference is attributed to C deposition in the outer reduced layers of the pellet, causing resistance to gas diffusion when the reducing gas contained CH4. It has been shown that the excess residual C can be removed from the reduced iron at lower temperature by its hydrogenation.
In another study, it has also been demonstrated that it is possible to hydrogenate residual C in direct reduced products to CH4. The C formed as a result of the reduction of Fe oxide in a mixture of CH4 and H2 (containing 20 % CH4) reacted with steam (H2O) according to the water gas reaction to regenerate H2 and produce CO.
Pure ferric oxide briquettes were reduced at temperatures ranging from 800 deg C to 1050 deg C, in gas mixtures containing H2, CO, CH4, N2 and CO2, which has been obtained by partial oxidation of natural gas with air. The CH4 content of the reformed gas mixture was between 13 % and 16 %. The overall reduction rate again has been controlled initially by chemical reaction and the gaseous diffusion has been applicable during the latter stages. It has been shown that the hematite ore briquettes have swelled and considerable porosity has been was developed during reduction. The solid-state diffusion rates increased more rapidly with temperature than it did by interfacial or gaseous diffusion reaction rates. The reduction of porous (30 % porosity) Fe ore in CH4 has indicated that the reaction proceeded stepwise from Fe2O3 to Fe3O4, FeO and Fe. The Fe catalyzed the CH4 cracking reaction. Optimum conditions for CH4 utilization occurred at around 1000 deg C.
The above findings are not consistent with the earlier studies on the understanding of high-grade porous (around 30 %) or dense hematite ore reduction kinetics, which had shown that the rate of reduction can be considered to fall between 3 limiting cases, namely (i) uniform internal reduction, (ii) limiting mixed control, and (iii) diffusion in porous iron layer, respectively with the rate of reduction corresponding to, (i) chemical control, (ii) the overall chemical control and diffusion control, and (iii) diffusion control. The overall rate of reduction is not controlled by only one of these rate controlling mechanisms and can be changed from one limiting case to another during the course of reduction.
In one of the studies it has been found that the most important factors controlling the extent of reduction are (i) the temperature, (ii) the composition of gas, presence of unreacted hydrocarbons in the reducing gas, the ratio of H2/C in it, and reducing capacity, (iii) the ore particle size, and (iv) the residence time for reduction.
Reduction of Fe oxides with the products of CH4 reformed with H, O within the reduction furnace – In early 1981 a commercial process has been introduced, using gaseous mixtures containing upto around 30 % by volume of CH4 (e.g. coke oven gas), for the direct gaseous reduction of Fe ore in a counter current moving bed shaft furnace. The furnace contained a reduction zone, a cooling zone, and an intermediate reforming zone. A hot mixture of coke oven gas and steam has been fed to the intermediate zone and reduced Fe ore therein catalyzed the reforming of the CH4 to CO and H2. The reformed gas flows upward into the reduction zone for the reduction of Fe ore.
제조공정
Energiron 직접 환원 기술 Energiron 직접 환원 기술은 가스 기반의 직접 환원 기술입니다. Energiron 공정은 철광석 알갱이 또는 덩어리를 금속 철로 변환합니다. Tenova와 Danieli가 공동으로 개발한 HYL 직접 환원 기술을 사용하며 액강 생산 비용을 낮추기 위한 경쟁력 있고 환경적으로 깨끗한 솔루션입니다. 간단한 플랜트 구성을 사용하고 다양한 환원 가스 소스를 사용할 수 있는 유연성이 있으며 철광석을 매우 효율적이고 유연하게 사용할 수 있습니다. 많은 공정 이점의 핵심 요소는 가압 작동과 직접적인
제철을 위한 Matmor 공정 Matmor 공정은 현재 Environmental Clean Technologies Ltd(ECT)에서 개발 중인 제철 공정입니다. Matmor 공정 기술은 특허 기술입니다. 이 기술은 갈탄을 기반으로 하며 독특한 화학 및 용광로 설계로 인해 고급 철광석을 저렴한 대체 원료로 대체할 수 있습니다. 일반적으로 갈탄(갈탄이라고도 함)은 휘발성 물질과 수분 함량이 높기 때문에 야금 용도로 사용되지 않습니다. Environmental Clean Technologies Ltd는 공장, 장비 및 지적 재산(