일반 기울기 근사 내의 모든 밀도 함수 이론 계산은 Vienna ab initio Simulation Package[35]를 사용하여 수행됩니다. 교환 및 상관 용어는 Perdew-Burke-Ernzerhof(PBE) 기능으로 설명되었으며 프로젝터 증강 파동 전위는 전자-이온 상호 작용을 설명하기 위해 사용되었습니다[36,37,38]. 도핑된 GeP3 단층은 32개의 원자를 포함하는 주기적인 2 × 2 슈퍼셀로 모델링되었으며, 3 × 3의 더 큰 슈퍼셀도 결과를 확인하는 데 사용되었습니다. z를 따라 약 20 Å의 진공 공간 인접 레이어 간의 상호 작용을 제거하기 위해 방향을 채택했습니다. 단일 도핑의 경우 하나의 Ge 또는 P 원자가 III족(IV, V 및 VI)의 도펀트로 대체되었습니다. 호스트 GeP3의 격자 상수 및 전자적 특성을 포함하여 보고된 결과와 비교하여 기하학적 구조가 결정됩니다. 단층. 도핑 시스템에서 슈퍼셀의 모든 원자는 Hellmann-Feynman 힘이 0.02 eVÅ
−1
미만이 될 때까지 이완됩니다. , 그러나 표면 셀의 격자 상수는 원자 이완 동안 고정됩니다. 약 600 eV 및 6 × 6 × 1 k의 운동 에너지 차단 -메쉬가 각각 사용되었습니다[39].
공동 도핑 시스템의 경우 유사한 공식이 사용됩니다.
$$ {\mathrm{E}}_{\mathrm{f}}\left(\mathrm{Ge}{\mathrm{P}}_3:\mathrm{XY}\right)=\mathrm{E}\left (\mathrm{Ge}{\mathrm{P}}_3:\mathrm{XY}\right)-\mathrm{E}\left(\mathrm{Ge}{\mathrm{P}}_3\right)-{ E}_{\mathrm{X}}-{E}_{\mathrm{Y}}+{E}_{\mathrm{i}}+{E}_{\mathrm{j}} $$ (2 ) 결과 및 토론
단일 원소 도핑 시스템을 위한 짝수-홀수 진동
그림 1a는 GeP3 구조의 평면도와 측면도를 보여줍니다. 2 × 2 슈퍼셀, 그림 1b는 GeP3의 해당 2D Brillouin 영역입니다. 단층. GeP3의 최적화된 격자 상수 단층은 \( \mathrm{a}=\mathrm{b}=6.96\ {\AA} \)이고 계산된 밴드갭은 약 0.26 eV로 다른 이론적인 계산과 잘 일치합니다.
<그림><그림><소스 유형="이미지/webp" srcset="//media.springerature.com/lw685/springer-static/image/art%3A10.1186%2Fs11671-019-3135-3/MediaObjects/ 11671_2019_3135_Fig1_HTML.png?as=webp"> 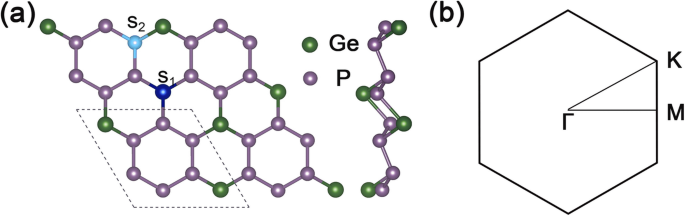
GeP3의 기하학적 구조와 브릴루앙 영역 . 아 GeP3의 최적화된 형상의 평면도 및 측면도 2 × 2 슈퍼셀. 점선은 GeP3의 단위 셀을 나타냅니다. 단층, S1 Ge 사이트의 대체 위치를 나타내며 S2 치환 위치 P 원자의 부위를 나타낸다. ㄴ GeP3의 2D 브릴루앙 구역 단층
먼저 단일 원소가 도핑된 GeP3의 밴드 구조를 플로팅했습니다. Ge 원자를 대체하는 단층(여기서 B, C, N, O, Al, Si, P, S, Ga, As 및 Se를 도펀트로 선택함). 결과는 각각 그림 2a-l에 나와 있습니다. 우리는 페르미 준위가 위로 이동하고 하나의 전자 도펀트로 인해 그룹 V(N, P, As)의 전도대를 가로지르는 반면, 그룹 III(B, Al, Ga) 도펀트는 하나의 전자가 적기 때문에 전도대를 가로지르는 것을 분명히 볼 수 있습니다. 하나는 아래로 이동하고 원자가 밴드를 교차합니다. 예를 들어, 그림 2f와 j에서 가전자대 최대값은 그림 2e와 i에 표시된 부분적으로 점유된 대역에 해당합니다. 그러나 그룹 IV(C, Si 및 Ge) 및 VI(O, S 및 Se) 도펀트의 경우 Ge 원자와 같거나 2개 더 많은 전자로 인해 시스템이 반도체 특성을 나타냅니다. 이러한 반도체에서 금속 전이로의 조정은 원자가 전자 수의 점유, 즉 홀수(짝수) 원자가 전자 점유가 금속(반도전) 특성으로 이어짐에서 비롯됩니다.
<그림><그림><소스 유형="이미지/webp" srcset="//media.springerature.com/lw685/springer-static/image/art%3A10.1186%2Fs11671-019-3135-3/MediaObjects/ 11671_2019_3135_Fig2_HTML.png?as=webp"> 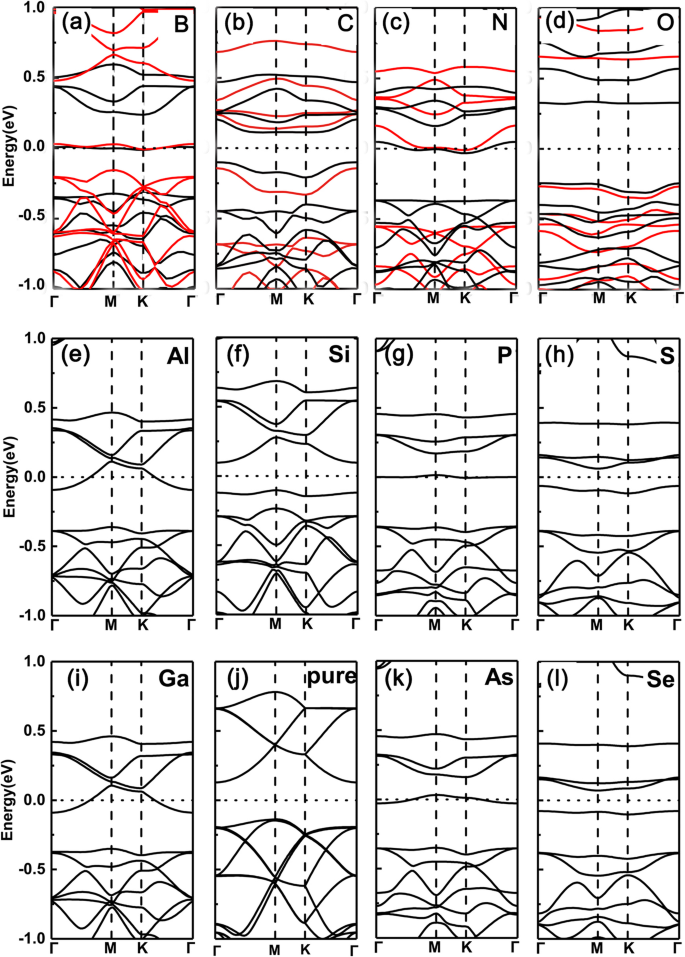
GeP3에서 다양한 도펀트의 밴드 구조 Ge 원자를 대체하는 단층. 아 나, 나 ㄷ, ㄷ N, d 오, 이 알, f 시, 지 피, h 에스, 나 가, j 순수 GeP3 , k l 세. GeP3의 다양한 도펀트가 있는 \( \mathsf{2}\times \mathsf{2} \) 슈퍼셀에 대한 계산된 밴드 구조 순수 GeP3와 함께 각각 Ge 원자로 치환된 III에서 VI까지의 단층 단층. PBE 및 HSE06 기능은 모두 최상단 행에 사용됩니다.
PBE 함수에서 파생된 위 결과의 유효성을 확인하기 위해 HSE06(하이브리드 밀도 함수) 함수를 사용하여 최상위 행의 도핑된 시스템도 확인합니다. PBE 기능은 실제로 과소 평가로 인한 밴드 갭의 실수를 제공합니다. 그러나 우리가 연구한 시스템에서는 모두 상당한 간격이 있습니다. 이는 PBE 기능으로 인한 금속성 또는 반도체 특성 간의 실수가 일반적으로 발생하지 않음을 의미합니다(이는 반도체의 일부 작은 밴드 갭에서 PBE 기능이 일반적으로 전도성과 금속성 특성 사이의 실수). 또한, 우리 연구에서 우리가 관심을 갖는 것은 밴드 갭의 특정 값 대신 금속 또는 반도체 특성입니다. PBE 기능에서 파생된 격차와 비교할 때 HSE06 기능에서 파생된 격차는 명확하게 확대됩니다. 이러한 경우에도 금속-반도체 진동은 그대로 유지됩니다. 따라서 PBE 기능을 기반으로 그린 핵심 성분은 신뢰할 수 있습니다.
그러나 뚜렷한 대조적으로 P 원자를 동일한 도펀트로 대체하는 경우는 각각 그림 3a-1에 표시된 것처럼 완전히 반대입니다. 즉, 그룹 V(N, As) 및 그룹 III(B, Al, Ga) 도펀트의 경우 도핑된 시스템은 반도체 특성을 유지하는 반면 그룹 IV(C, Si, Ge) 및 VI(O, S, Se)의 경우 도펀트는 금속 기능으로 변경됩니다(여기서 PBE와 HSE06 기능 간에도 동일한 경향이 나타납니다). 이는 원자가 전자가 고유 GeP3와(보다) 동일하게(2개 적음) 유지되기 때문입니다. 그룹 V(그룹 III) 도펀트의 경우 그룹 IV(VI) 도펀트의 경우 전자가 하나 더 적습니다.
<그림><그림><소스 유형="이미지/webp" srcset="//media.springerature.com/lw685/springer-static/image/art%3A10.1186%2Fs11671-019-3135-3/MediaObjects/ 11671_2019_3135_Fig3_HTML.png?as=webp"> 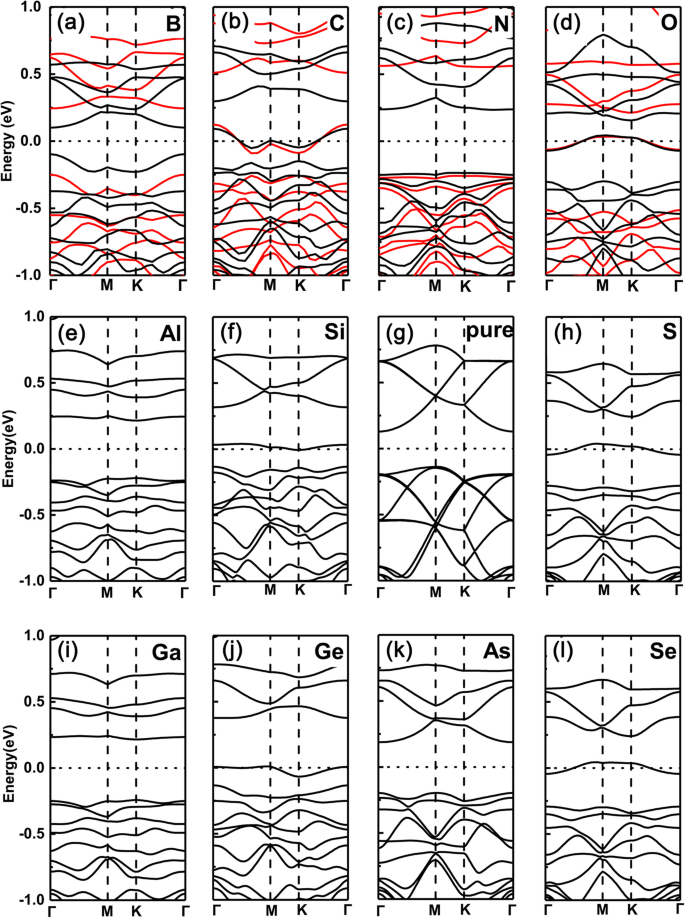
GeP3에서 다양한 도펀트의 밴드 구조 치환된 P 원자를 갖는 단층. 아 나, 나 ㄷ, ㄷ N, d 오, 이 알, f 시, 지 피, h 에스, 나 가, j 순수 GeP3 , k l 세. GeP3의 다양한 도펀트가 있는 \( \mathsf{2}\times \mathsf{2} \) 슈퍼셀에 대한 계산된 밴드 구조 순수 GeP3와 함께 각각 P 원자로 치환된 III에서 VI까지의 단층 단층. PBE 및 HSE06 기능은 모두 최상단 행에 사용됩니다.
반도체에서 금속 특성으로의 전이 진동을 더 잘 나타내기 위해 우리는 각각 그림 4a와 b에 표시된 것처럼 서로 다른 도펀트에 따라 밴드 갭의 변화 추세를 플로팅했습니다. 분명히 우리는 반도체에서 금속 속성으로의 전환이 급격하게 역전되고 있음을 알 수 있습니다. 구체적으로, 금속성(반도성)-반도성(금속성) 진동은 III족에서 VI족에 이르는 도펀트로 Ge(P) 자리를 대체할 때 발생합니다. 또한 밴드갭의 크기가 고유 GeP3와 거의 동일하게 유지된다는 흥미로운 현상도 발견했습니다. 도펀트가 Ge 원자와 동일한 원자가 전자를 가질 때 단층. 그러나 도펀트가 Ge 원자보다 전자를 2개 더 많이 가질 때 밴드 갭의 크기가 상대적으로 크게 변합니다. 그럼에도 불구하고 P 사이트의 도펀트에 대해서는 원자가 전자의 수에 관계없이 밴드 갭의 크기가 항상 상대적으로 크게 변합니다. 이것은 원자의 반경과 사용 가능한 원자가 전자의 결합 효과, 즉 Ge 원자와 거의 동일한(작거나 더 큰) 반경 및 원자가 전자를 갖는 도펀트가 전자에 상대적으로 더 작은(더 큰) 효과를 일으키는 것으로 이해될 수 있습니다. 밴드 갭과 같은 속성. 이것은 반도체-금속 전이의 진동을 조정할 수 있을 뿐만 아니라 적절한 도펀트와 다른 도핑 사이트를 선택하여 밴드 갭의 크기를 조정할 수 있음을 의미합니다.
<그림><그림><소스 유형="이미지/webp" srcset="//media.springerature.com/lw685/springer-static/image/art%3A10.1186%2Fs11671-019-3135-3/MediaObjects/ 11671_2019_3135_Fig4_HTML.png?as=webp"> 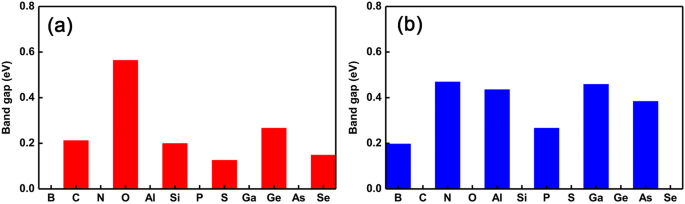
모든 단일 도핑 시스템의 밴드 갭. 도핑된 GeP3의 밴드 갭 그룹 V에서 VI에 이르는 다양한 도펀트를 가진 단층. 아 Ge 원자 및 b의 치환 치환 P 원자, 각각
GeP3에서 서로 다른 도펀트의 전자 구조 변화를 이해하기 위해 단층, 우리는 GeP3에서 고유 및 도핑 그룹 IV–V의 부분 밀도 상태(PDOS)를 플로팅했습니다. 각각 그림 5a-d와 같이 단층. GeP3의 가전자대 최대값(VBM)과 전도대 최소값(CBM)이 단층은 주로 Ge와 P 원자의 p 오비탈에서 유래합니다. C 및 Si와 같이 Ge 원자와 동일한 수의 원자가 전자를 가진 도펀트를 사용할 수 있는 경우 고유 GeP3의 VBM 바로 위에 위치하는 불순물 상태가 있습니다. C와 Si의 p 궤도 에너지 준위가 P 원자의 p 궤도 에너지 준위보다 높기 때문입니다(그림 5a 참조). 따라서 전도성이 그대로 유지되고 밴드갭 변화의 크기가 상대적으로 작다. 그러나 도펀트가 N, P 및 As와 같이 Ge 원자보다 하나 이상의 전자를 가질 때 밴드 갭에도 불순물 상태가 있고 불순물 상태는 분열하는 CBM(dominant)과 상태의 혼성화에서 비롯됩니다. 도펀트(그림 5b 참조).
<그림><그림><소스 유형="이미지/webp" srcset="//media.springerature.com/lw685/springer-static/image/art%3A10.1186%2Fs11671-019-3135-3/MediaObjects/ 11671_2019_3135_Fig5_HTML.png?as=webp"> 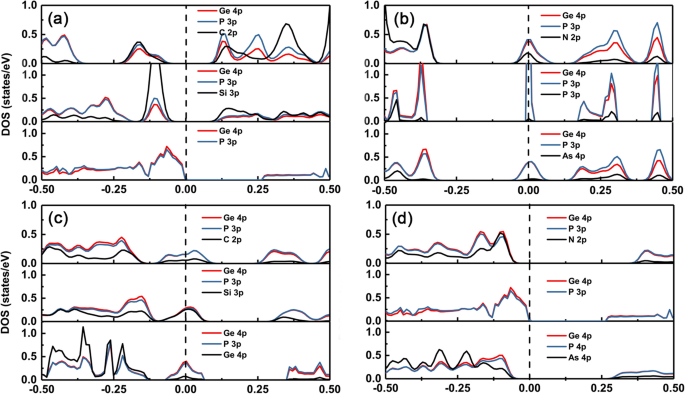
도핑된 시스템용 DOS. GeP3 도핑된 IV족(C, Si, Ge) 및 V족(N, P 및 As) 원자에 대한 상태의 부분 밀도(오른쪽) . 검은색 세로 점선은 페르미 준위입니다. (a) 및 (b) 치환된 Ge 원자, (c) 및 (d) 치환된 P 원자
반대로 P 사이트에 도핑하는 경우 IV족과 같이 도펀트가 P 원자보다 원자가 전자가 하나 적은 경우 페르미 준위 전체에 불순물 상태가 있게 되고 불순물 상태는 분할 VBM(dominant ) 및 도펀트의 상태. 반면, 도펀트가 V족과 같은 P 원자와 동일한 수의 원자가 전자를 가질 때 도핑된 시스템은 여전히 반도체 특성을 유지합니다(그림 5c 참조). 밴드 갭은 고유 GeP3의 밴드 갭보다 상대적으로 커집니다. 격자 상수의 더 큰 불일치로 인한 단층. 더욱이, 우리는 또한 P 원자를 치환할 때 N 도펀트의 밴드 갭이 As 도펀트의 밴드 갭보다 더 크다는 것을 관찰했다. 이것은 As 원자의 p 궤도 에너지 준위가 N 원자의 에너지 준위보다 높기 때문에 p 궤도 에너지 준위가 높을수록 불순물 상태가 VBM에서 더 많이 위쪽으로 이동하기 때문입니다(그림 5d 참조).
공동 도핑 시스템에 대한 논리적 관계
따라서 단일 서로 다른 도펀트에 대한 앞에서 언급한 발견을 기반으로 우리가 원하는 전자 특성을 충족하는 공동 도핑 시스템을 설계할 수 있습니다. 여기에서는 공동 도핑 효과를 설명하기 위한 예로 B, C, N 및 O의 결과만 보여주지만 결론은 서로 다른 선택된 도펀트에 대해 강력합니다. 예를 들어, Ge 사이트 공동 도핑에서 원자가 전자가 하나 더 적은 두 도펀트 모두 자연적으로 반도체 특성을 유발할 수 있는 반면, 원자가 전자가 하나 더 적은 두 도펀트의 경우 공동 도핑 시스템은 따라서 또한 다음과 같이 할 수 있습니다. 반도체 속성이 있습니다.
그러나, 하나 더 적은(더) 그리고 동일한 원자가 전자를 갖는 두 개의 도펀트의 경우, 공동 도핑 시스템은 하나의 더 적은 수의 원자가 전자 도펀트가 특성을 생성하므로 금속 특성을 여전히 유지합니다. 단순화, 이 아이디어는 공동 도핑된 시스템의 추가 밀도 기능 이론(DFT) 계산에 의해 정확하게 확인됩니다. B, C, N 및 O 공동 도핑된 GeP<하위>3 단층.
<그림><그림><소스 유형="이미지/webp" srcset="//media.springerature.com/lw685/springer-static/image/art%3A10.1186%2Fs11671-019-3135-3/MediaObjects/ 11671_2019_3135_Fig6_HTML.png?as=webp"> 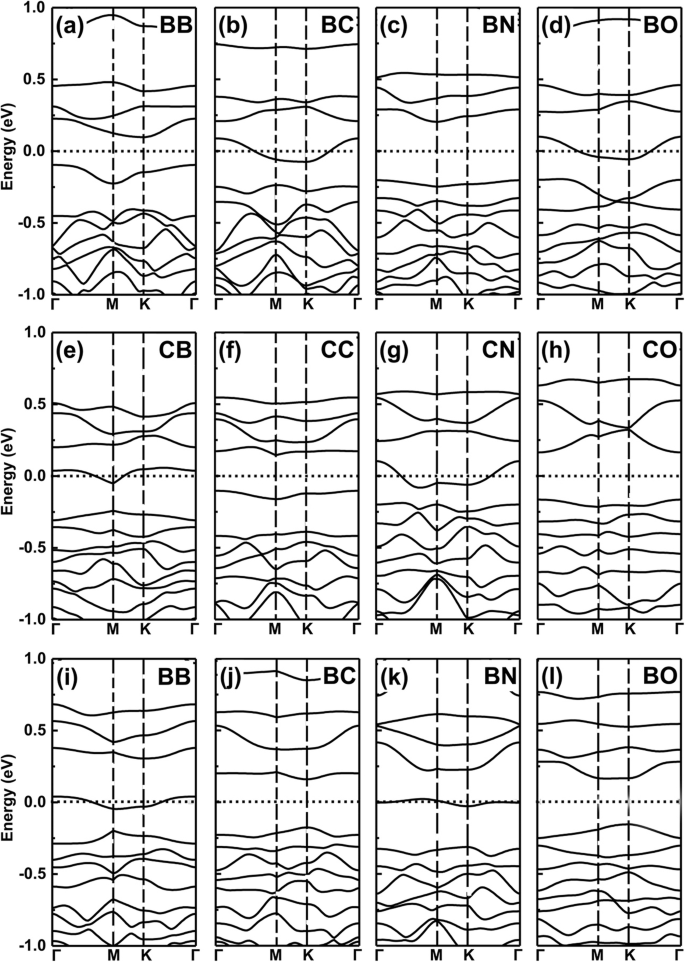
공동 도핑 시스템의 밴드 구조. B, C, N 및 O 공동 도핑된 GeP3의 밴드 구조 단층. 아 –d 2개의 도펀트는 GeP3에서 2개의 Ge 원자를 대체합니다. 단층, e –h 두 개의 도펀트는 두 개의 P 원자를 대체합니다. i –나 두 개의 도펀트가 각각 하나의 Ge 원자와 하나의 P 원자를 대체합니다.
이제 금속 특성을 "M"으로 설정하고 반도체 특성을 "S"로 설정하여 논리 유사 연산 "AND"를 제공할 수 있습니다. 우리는 각각 M AND M =S, S AND S =S, M AND S =M과 같은 논리적 관계를 정의합니다. 여기에서 우리가 위에서 얻은 이러한 발견은 논리와 유사한 관계를 따릅니다. 예를 들어, 원자가 전자가 하나 더 많고 더 적은 도펀트는 금속 특성을 나타내지만, B 및 N과 같은 공동 도핑으로 두 도펀트를 사용할 때 그림 7a 및 b에 표시된 것처럼 Ge 사이트에서 공동 도핑된 시스템은 그림 6c를 참조하여 예상한 대로 반도체 특성이 됩니다. B-C 공동 도핑된 GeP3를 선택하면 단층 시스템에서는 M AND S의 경우인 금속 특성을 나타냅니다(그림 4a, b 참조). GeP3,에 공동 도핑된 C-N, N-O 및 B-O 원자에 대해 동일 2개의 Ge 원자, 2개의 P 원자, 또는 1개의 Ge 및 P 원자를 각각 그림 7c-f에 표시된 대로 대체합니다.
<그림><그림><소스 유형="이미지/webp" srcset="//media.springerature.com/lw685/springer-static/image/art%3A10.1186%2Fs11671-019-3135-3/MediaObjects/ 11671_2019_3135_Fig7_HTML.png?as=webp"> 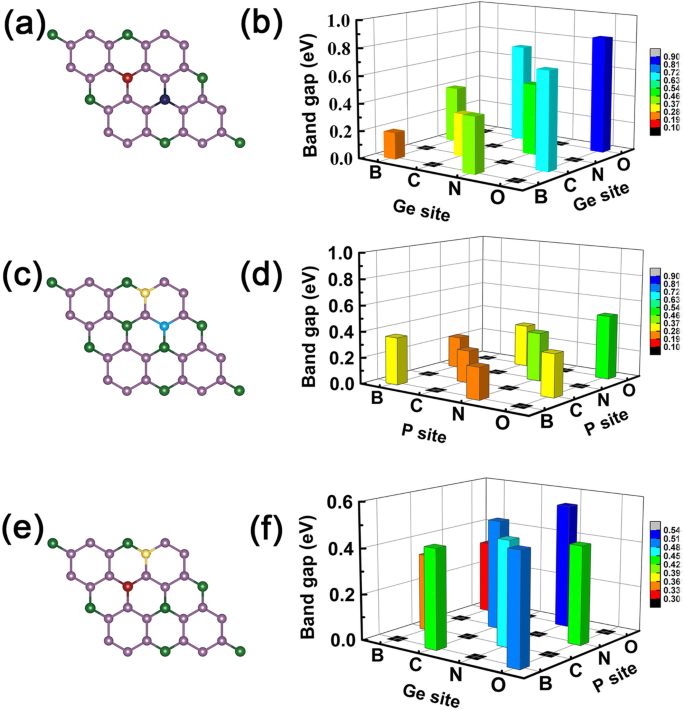
모든 공동 도핑 시스템의 밴드 갭. 공동 도핑된 GeP3의 밴드 갭 크기 monolayer에서 왼쪽은 co-doped site의 스케치이고 오른쪽은 doping element에 해당하는 band gap의 크기이다. 아 , b 원소를 도핑하는 경우는 두 개의 Ge 원자를 차지합니다. ㄷ , d 원소를 도핑하는 경우는 두 개의 P 원자를 차지합니다. 이 , f 원소를 도핑하는 경우는 각각 Ge 및 P 원자를 차지합니다.
마지막으로 단일 도핑 시스템과 공동 도핑 시스템 모두의 안정성을 확인하여 실험에서 추가로 실현할 수 있는지 확인했습니다. 형성 에너지는 식을 사용하여 계산됩니다. (1) 및 (2)는 각각 단일 도핑 및 공동 도핑의 경우입니다. 결과는 그림 8a-e에 나와 있습니다. 그림 8a와 b에서 E f Ge 사이트에서 단일 도펀트의 비율은 모두 GeP3의 단일 도펀트에 가깝습니다. 단층(기준점으로 0으로 설정), C, N 및 S 원자 제외. 우리는 또한 B, O, P, Ge 및 Se 원자의 도펀트의 경우 형성 에너지가 다른 도펀트보다 훨씬 작아 실험에서 도핑하기가 매우 쉽다는 것을 알았습니다. P 사이트의 도펀트의 경우 B, O, P 및 Ge 원자의 도펀트가 상대적으로 형성 에너지가 작고 도핑이 용이합니다. C, N, Al, Ga는 도핑이 쉽지 않습니다.
<그림><그림><소스 유형="이미지/webp" srcset="//media.springerature.com/lw685/springer-static/image/art%3A10.1186%2Fs11671-019-3135-3/MediaObjects/ 11671_2019_3135_Fig8_HTML.png?as=webp"> 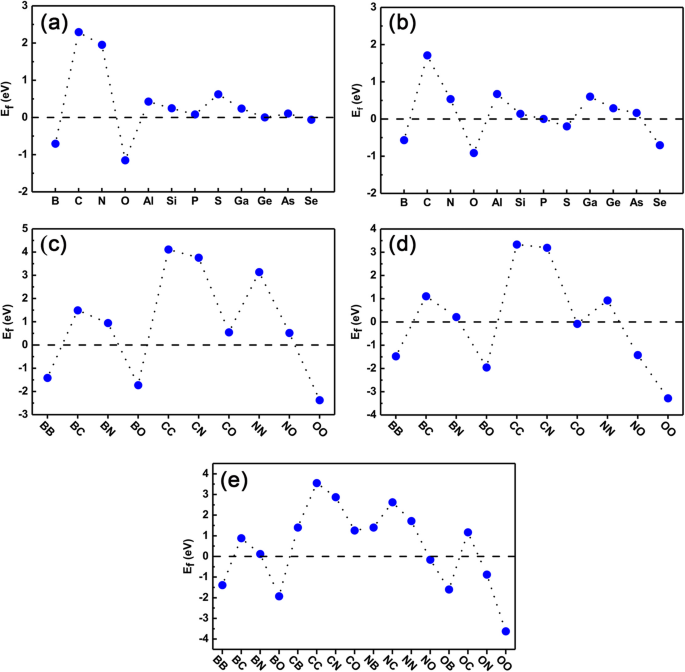
모든 도핑된 시스템의 형성 에너지. 단일 원소 도핑 및 공동 도핑 시스템의 계산된 형성 에너지. 아 , b 는 각각 도펀트 치환된 Ge 원자 및 P 원자이고; ㄷ –이 2개의 Ge 원자, 2개의 P 원자, 1개의 Ge 원자 및 1개의 P 원자로 각각 치환된 공동 도펀트
공동 도핑의 형성 에너지에 관하여, 그림 8c-e는 두 개의 도펀트가 두 개의 Ge 원자(Ge-Ge 사이트로 표시됨), 두 개의 P 원자(PP로 표시됨)의 위치를 차지하는 공동 도핑의 형성 에너지입니다. 사이트), 각각 하나의 Ge 및 하나의 P 원자(Ge-P 사이트로 표시됨). 여기에서는 B, C, N 및 O의 도펀트 결과만 예시로 보여줍니다. Ge-Ge 사이트 및 P-P 사이트의 경우 공동 도핑의 형성 에너지는 단일 원소 도핑의 형성 에너지를 개별적으로 평균화하여 대략적으로 추정할 수 있습니다. 분명히, Ge-Ge 사이트에서 BB, BO 및 OO 공동 도핑 및 P-P 사이트에서 BB, BO, NO 및 OO 공동 도핑의 경우 형성 에너지가 상대적으로 작고 실험에서 쉽게 실현될 수 있습니다. 그러나 Ge-Ge 사이트에서 CC, CN 및 NN 공동 도핑 및 P-P 사이트에서 CC 및 CN 공동 도핑의 경우 형성 에너지가 상대적으로 커서 실험에서 도핑하기 어렵다는 것을 나타냅니다. Ge-P 사이트 공동 도핑의 경우, 도 8e에 도시된 바와 같이, 형성 에너지는 도펀트 사이에 전하 이동이 있기 때문에 Ge-Ge 또는 P-P 사이트 공동 도핑보다 더 복잡해진다. 어쨌든 BB, BO 및 OO 동시 도핑은 형성 에너지가 더 작은 반면 CC, CN 및 NN 동시 도핑은 더 큰 형성 에너지를 갖습니다. 일반적으로 형성 에너지는 도펀트의 원자가 전자 수에 크게 의존한다. 구체적으로, 치환된 원자보다 전자가 하나 적은(더) 두 개의 도펀트가 있는 경우, 동시 도핑된 시스템의 형성 에너지는 BB(NN) 동시 도핑된 Ge 사이트와 같은 대응하는 단일 도펀트의 형성 에너지보다 낮습니다(높음). 이것은 도핑된 시스템의 감소(증가) 전자의 감소(증가) 에너지와 쿨롱 반발 사이에 경쟁이 있기 때문입니다. 정공-정공 동시 도핑의 경우 전자의 경우 에너지가 후자의 경우보다 훨씬 크므로 BB와 같은 동시 도핑 시스템에서 형성 에너지가 상당히 감소하는 반면 전자-전자 동시 도핑의 경우 둘 다 전자와 나중의 경우는 NN과 같은 더 높은 형성 에너지로 이어집니다. 그러나 BN 동시 도핑된 Ge 사이트와 같은 정공 및 전자 동시 도핑 시스템의 경우 형성 에너지는 대응하는 단일 도핑된 경우보다 극적으로 낮습니다. 이는 이러한 공동 도핑된 시스템에서 시스템에 순 첨가 또는 환원된 전자로부터 에너지 이득이 없고 쿨롱 상호작용이 공동 도핑된 도펀트를 형성하는 데 결정적인 역할을 하기 때문입니다. 전체적으로 블랙 포스포렌의 원소 도핑에 대한 이전 연구를 종합하면 현재 연구는 어느 정도 보편성을 가지며 BN, MoS2와 같은 다른 2D 반도체 단분자막을 적용할 것으로 예상된다는 점을 지적해야 합니다. , 등등.
마지막으로 도핑되지 않은 경우와 비교하여 위의 도핑된 시스템의 안정성을 확인하기 위해 그림 9a-f와 같이 에너지 대 시간을 표시하기 위해 AIMD(초기 분자 역학)를 수행했습니다. 시간이 충분히 지속되는 한(~ 4 ps) 진동 진폭이 수렴한다는 것을 분명히 알 수 있습니다. 이는 도핑된 시스템이 C-도핑된 GeP3 도 9a에서. 가장 활동적인 C 원자 도핑 GeP3의 경우에도 , 극한 온도는 그림 9c와 같이 최대 300 K일 수 있습니다. 또한 Ge 사이트에서 금속 Al 치환을 예로 들어 계산된 결과를 그림 9e 및 f에 표시합니다. 이로부터 에너지 진동 진폭이 시간이 지남에 따라 점진적으로 감소한다는 것을 분명히 알 수 있습니다. 즉, 에너지가 시간이 충분히 지속되고 도핑된 시스템의 구조가 열 변동에 대해 열적으로 안정되는 한 수렴해야 합니다. 따라서 우리는 고품질 GeP3를 감안할 때 이러한 도핑된 시스템이 추가 실험에서 실현될 수 있다고 기대할 수 있습니다. 단층이 준비되었습니다.
<그림><그림><소스 유형="이미지/webp" srcset="//media.springerature.com/lw685/springer-static/image/art%3A10.1186%2Fs11671-019-3135-3/MediaObjects/ 11671_2019_3135_Fig9_HTML.png?as=webp"> 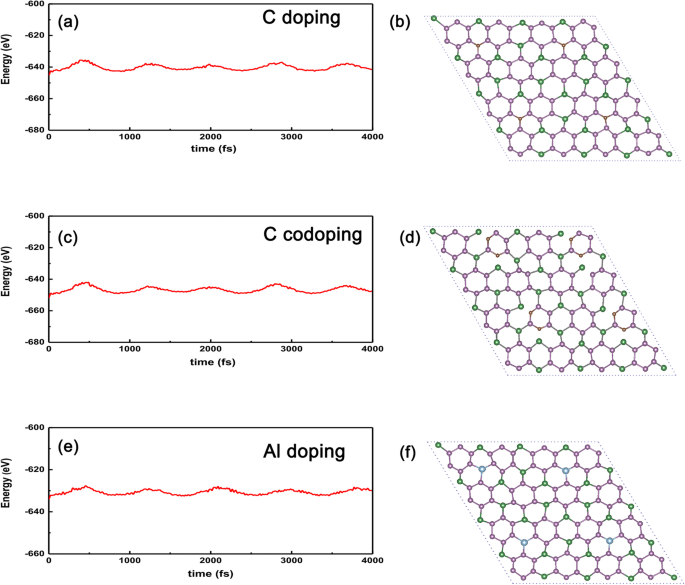
C, 2개의 C 및 Al 원자 도핑 시스템용 AIMD. AIMD는 a의 열 안정성을 확인합니다. C 원자 도핑 GeP3 Ge 원자를 대체하여 c 두 개의 C 원자 도핑된 GeP3 2개의 P 원자로 치환, 및 e Al 원자 도핑된 GeP3 300 K에서 치환으로. b의 구조 C, d 두 개의 C 원자 및 f Al은 4000 fs 이후의 최종 구조에 해당합니다.
마지막으로 여기에 제시된 연구의 신뢰성에 대해 논의하고자 합니다. 여기에 제시된 우리의 결론은 이론적으로 예측된 결과이지만 매우 신뢰할 수 있습니다. 이것은 여기에 사용된 호스트 재료가 보고되었고 계층화된 GeP3의 벌크 단계 때문입니다. 이미 존재한다[26]. 따라서 우리가 연구한 도핑 유도 관련 현상은 일단 단층 GeP3 더욱 실현됩니다. 그런 다음 해당 원자를 도핑할 수 있습니다. 단순화를 위해 단층 GeP3에 전자 또는 정공 도핑 일부 분자의 흡착에 의해 실현될 수 있습니다.
결론
요약하면, 우리는 2D GeP3에서 III 내지 VI족 도펀트의 전자적 특성을 조사했습니다. 단층 및 도핑된 GeP3 Ge 사이트의 치환으로 금속-반도체 진동은 도펀트의 원자가 전자 수의 함수로 나타나는 반면 이러한 진동은 P 사이트에서 치환으로 반전됩니다. 단일 도펀트의 결과를 기반으로 GeP3에서 공동 도핑의 전도성 특성을 제안할 수 있습니다. , 이는 간단한 논리 연산으로 얻을 수 있습니다. 마지막으로 다양한 도펀트의 형성 에너지를 계산하고 특히 전자-정공 및 정공-정공 동시 도핑의 경우 일부 동시 도핑 시스템이 쿨롱 인력으로 인해 에너지적으로 더 유리하다는 것을 발견했습니다. 우리의 발견은 새로운 현상을 제시할 뿐만 아니라 2D 이진 반도체에서 전자 특성을 조정하는 흥미로운 경로를 제안합니다.
데이터 및 자료의 가용성
현재 연구 중에 생성 및/또는 분석된 데이터 세트는 요청 시 해당 저자에게 제공됩니다.
약어
- 1D:
-
1차원
- 2D:
-
2차원
- AIMD:
-
Ab 초기 분자 역학
- BP:
-
블랙 포스포린
- CBM:
-
전도 대역 최소
- DFT:
-
밀도 함수 이론
- HSE06:
-
하이브리드 밀도 기능
- PBE:
-
퍼듀-버크-에른처호프
- PDOS:
-
상태의 부분 밀도
- VBM:
-
가전자대 최대값


