대부분의 알츠하이머병 약물은 혈액-뇌 장벽 때문에 효율적으로 작동하지 않습니다. 따라서 우리는 뇌 조직 전달을 실현하기 위한 도네페질(DZP) 운반체로서 폴리소르베이트 80(PS) 표면 적용 범위를 갖는 콜레스테롤 변형 풀루란(CHP) 나노입자(PS-DZP-CHP)를 설계했습니다. 크기 분석 및 등온 적정 열량 측정법을 통해 나노 물질과 약물의 최적 투여 비율(1:5)을 선택하고 나노 입자의 효능을 검증하기 위한 일련의 실험을 설계했습니다. 시험관 내 방출 실험의 결과에 따르면 나노입자는 72시간 이내에 지속적인 약물 방출을 달성할 수 있습니다. 마우스의 형광 관찰 결과 PS-DZP-CHP 나노 입자의 우수한 뇌 표적화를 보여주었다. 또한, 나노 입자는 마우스의 뇌 조직 농도에서 약물을 향상시킬 수 있습니다. DZP-CHP 나노 입자는 Aβ 단백질 손상으로 신경 세포를 전처리하는 데 사용되었습니다. 젖산 탈수소효소의 농도는 MTT, 로다민 123 및 AO-EB 염색에 의해 결정되었으며, 이는 DZP-CHP 나노입자가 Aβ25-35에 의해 유도된 신경독성에 보호 효과가 있음을 입증했습니다. 무료 도네페질보다 우수했습니다. Microthermal perpetual motion meter 테스트는 PS-DZP-CHP 나노입자가 뇌 조직을 표적으로 하는 나노입자에 필수적인 아포지단백 E와 친화력이 있음을 보여주었습니다.
소개
알츠하이머병은 진행성 인지기능장애를 일으키는 복잡한 병리학적 기전을 가진 중추신경계 질환이나 약물 투여의 어려움과 뇌조직에 도달할 수 있는 약물의 농도가 낮아 치료가 매우 어려운 질환이다[1, 2]. BBB의 통과는 약물 전달 시스템(DDS) 연구에서 주요 어려움이 되었습니다[3, 4]. 대부분의 약물은 생물학적, 화학적 또는 물리적 수단을 통해 BBB를 엽니다. 그 중 현재 임상적으로 가장 많이 사용되는 방법은 물리적인 방법이다[5]. 침습적 기술에 기반한 두개내 약물 전달의 극복할 수 없는 결함으로 인해 약물 표면의 화학적 변형을 통해 친유성 전구약물 또는 능동 수송 기질을 생산하는 것이 더 많은 이점이 있습니다[6]. 그 중 나노 입자는 BBB를 적극적으로 표적화하여 두개내 약물 전달을 달성하는 최선의 선택 중 하나입니다[7,8,9].
나노-DDS는 뇌 표적 연구의 초점이 되었으며 치료 및 진단을 제공하도록 설계된 폴리머 나노입자, 유기 나노입자, 리포솜, 나노섬유 및 미셀을 포함합니다[6, 10,11,12]. 약물이 장착된 나노입자와 소분자 친유성 약물은 모두 BBB를 통과할 수 있습니다. 차이점은 약물이 탑재된 나노입자가 뇌의 모세혈관 벽에 흡착되어 수동 확산을 통과할 가능성이 더 높다는 것입니다. 또한, 자유 약물은 두 번째 장벽인 혈액-뇌척수액 장벽(B-CSF)에 직면합니다[13]. 다른 장벽 대응물과 달리 대부분의 약물은 CSF를 통해 비교적 투과성이고 뇌 실질로 확산됩니다. 그러나 매우 느린 확산 과정으로 인해 CSF의 유리 약물 농도는 뇌 실질보다 훨씬 높으며 CSF의 높은 농도는 약간의 독성을 유발할 것입니다 [14, 15]. 이 과정에서 나노 입자는 독특하고 중요한 이점을 가지고 있습니다. 나노캐리어의 표면이 친수성 계면활성제로 코팅되면 ApoE가 표면에 흡착되고 CSF는 나노입자가 혈관주변 공간을 통해 뇌실질로 이동하는 것을 촉진할 수 있다[16]. Kreuter 등에 의해 제조된 비이온성 계면활성제 폴리소르베이트 80으로 코팅된 나노입자. [17]은 뇌 시스템에 성공적으로 전달되고 혈장에서 혈청 단백질 ApoE의 흡착을 통해 수송된 최초의 약물이었습니다. 따라서 나노입자 표면의 폴리소르베이트 80 변형을 통해 약물 특이적 복합체를 형성하면 약물 표적화 및 내인성 BBB 수용체 인식이 가능해지며 약물의 생체 이용률이 크게 증가합니다[18,19,20].
현재, 아세틸콜린에스테라제(AChE) 억제제 도네페질(DZP)은 중등도 알츠하이머병 치료에 일반적으로 사용되지만 지용성으로 인해 생체에서 잘 용해되지 않습니다. , 경구 생체 이용률이 낮기 때문에 기존의 도네페질 정제를 매일 복용해야 치료 효과를 유지할 수 있습니다[21, 22]. 알츠하이머병 환자의 인지 능력은 심각하게 손상되어 복약 일정을 유지하는 데 큰 불편을 겪기 때문에 지속형 서방성 DZP 제제의 개발이 시급하다. 아밀로이드 캐스케이드 가설은 알츠하이머병의 원인이 뇌실질과 뇌혈관벽 주위에 아밀로이드 플라크의 침착이라는 것을 시사합니다. 무정형의 고도로 섬유성이며 불용성인 세포외 Aβ 침착물로 구성된 뇌 영역에서도 다수의 확산 반점이 관찰됩니다[23, 24]. Boridy et al. 무독성이며 쉽게 분해되는 풀루란 다당류 나노입자는 Aβ 단백질과 복합체를 형성하여 단백질 응집을 효과적으로 방지하고 세포에서 빠르게 제거되어 세포 독성을 억제할 수 있음을 발견했습니다[25]. CHP(Cholesterol-hydrophobicically Modified pullulan)는 수용액에서 소수성 코어와 당 사슬 친수성 껍질을 가진 나노구조로 자가 조립될 수 있는 양친매성 물질이다[26, 27]. 동시에, CHP 나노입자는 Aβ 단백질을 흡착하여 그의 침착 및 응집을 방지하여 상승적인 역할을 할 수 있다. 나노운반체로서 CHP는 상당한 우월성을 가지고 있습니다.
위의 지식을 바탕으로 연구팀은 DZP-CHP 나노 제제를 초기에 설계하고 나노캐리어에 대한 약물의 최적 투여 비율을 결정했다. 그런 다음, 폴리소르베이트가 나노입자의 표면에 흡착되어 BBB를 통한 통로를 능동적으로 표적화하여 뇌 농축을 달성했습니다. 이 연구에서 일련의 DZP-CHP 나노 용액을 특성화하고 시험관 내 약물 방출 과정을 조사하고 연구했습니다. 나노 입자가 성공적으로 준비된 후 Aβ25-35를 사용하여 신경 세포 손상을 유도하여 AD 세포 모델을 확립했습니다[28, 29]. 그런 다음 DZP-CHP 나노 용액의 보호 효과를 PC12 및 SH-SY5Y 세포 모델에서 조사했습니다.
자료 및 방법
자료
다음이 사용되었습니다. 콜레스테롤 소수성으로 변형된 풀루란(수제) [30]; 도네페질(Shanghai Ziqi Biotechnology Co., Ltd.); 폴리소르베이트 80(Tianjin Fuchen Reagent Institute); 인도시아닌 그린(ICG) 염료(Tianjin Baiying Biological Technology Co., Ltd.); 폴리소르베이트 80(Tween 80, PS)(Tianjin Fuchen Reagent Office); 검은 쥐(Hunan Slake Jingda Laboratory Animal Co., Ltd.); Aβ25-35(US Sigma); 테트라메틸 아조졸 염(MTT)(US Sigma); 신생아 소 혈청(U.S. Gibco); 젖산 탈수소효소 키트(LDH)(Nanjing Jiancheng Biological Co., Ltd.); AO/EB 이중 염색 형광 키트(Sino Pharmaceutical Group Chemical Reagent Co., Ltd.); 및 Central South University의 Second Xiangya Hospital 신경과에서 얻은 PC12 세포(쥐 부신 갈색세포종 세포). SH-SY5Y 세포(인간 골수 신경모세포종 세포)는 ATCC Cell Bank(Manassas, VA, USA)에서 구입했습니다.
나노입자의 준비
DZP 및 CHP 비율(w/w)(10:20, 4:20 및 2:20)이 서로 다른 세 가지 유형의 나노입자가 문헌에 보고된 방법에 따라 물 투석을 통해 먼저 성공적으로 준비되었습니다[31]. 일정 농도의 DZP-CHP 나노입자를 10mL의 일정한 부피로 비이커에 첨가한 다음, 폴리소르베이트 80(PS) 유화제(농도 0.7mmol)를 포함하는 다른 비커에 흡입하여 1시간 동안 침전시켰다. 그런 다음 혼합물을 EP 튜브에 넣고 3분 동안 초음파 처리했습니다(출력 전력 100W, 간헐적 펄스 작동 모드:펄스 폭 2.0초, 간헐적 시간 2.0초). 균일한 분산이 얻어질 때까지 작업을 3회 반복하였다[32]. 여과를 통해 불순물을 제거한 후 최종적으로 폴리소르베이트 80 유화 도네페질 약물 로딩 나노입자(PS-DZP-CHP)를 얻었다.
나노 입자의 특성
나노입자 형태
DZP 대 CHP 비율이 1:2, 1:5 및 1:10인 DZP-CHP 나노입자(DCP)의 모양, 표면 형태 및 크기를 Tecnai F20 투과 전자 현미경으로 분석했습니다. CHP, DZP-CHP 및 PS-DZP-CHP 나노입자 한 방울을 탄소가 코팅된 구리 메쉬에 놓아 얇은 액체 필름을 형성했습니다. 그 다음, 2%(w/v) 인텅스텐산 용액을 사용하여 필름의 자연 건조 후 샘플의 음성 염색을 얻었다. 새로 준비한 나노입자 수용액을 깨끗한 실리콘 웨이퍼에 적가하고 상온에서 건조시킨 후 JSM-6700F 전계방출 주사형 전자현미경으로 표면 구조를 관찰하였다.
나노입자 크기 및 제타 전위
DZP-CHP 및 PS-DZP-CHP 나노 입자의 크기, 다분산 계수(PDI) 및 제타 전위를 동적 광산란(DLS)을 사용하여 분석했습니다. 얻어진 균일 현탁액의 평균 입도 및 입도 분포를 각각 3회 측정하였다.
체외 약물 방출
도네페질의 방출은 동적 물 투석을 사용하여 측정되었습니다. DZP-CHP 및 PS-DZP-CHP 나노입자 1mg을 인산완충식염수(PBS, pH 7.4, 농도 0.01M) 5ml에 녹인 다음 투석백에 옮겨 동일한 용액에 100℃에서 보관했습니다. 자기 교반으로 37°C의 일정한 온도. 0, 0.5, 1, 2, 4, 8, 12, 24, 48, 72시간에 4밀리리터의 PBS를 동일한 pH에서 동일한 부피의 PBS로 희석했습니다. 자외선-가시광선 분광광도법을 사용하여 서로 다른 시간에 312nm에서 투석액의 흡광도를 감지했습니다. 용액의 함량은 표준곡선으로 결정하였고, 방출시험은 시험관내에서 3회 반복하였다. 도네페질의 방출 백분율은 다음 공식에 따라 계산되었습니다.
Cn은 Tn 시점의 샘플 농도(μg/mL)입니다. V는 PBS 방출 용액의 총 부피, mL이고; Vn은 Ti 시점에서의 PBS 방출 액체 부피, mL이고; Ci는 Ti 시점에서의 도네페질 농도, μg/mL입니다.
등온 적정 열량계(ITC)
CHP 나노입자 용액에 일정 농도의 PS 용액을 적하하고 ITC(vip-itc, Microcal, Northampton, MA, USA)로 열 변화를 측정하였다. CHP 나노입자 용액은 DZP 대 CHP 비율(1:2, 1:5 및 1:10)이 서로 다른 세 가지 유형의 나노입자를 포함했습니다. 모든 용액은 적정 전에 탈기되었습니다. 전체 시스템의 온도는 25°C로 일정하게 유지되었습니다.
뇌 표적을 관찰하기 위한 동물 실험
ICG 표지 도네페질 CHP 나노입자의 제조
CHP-DZP 400mg과 ICG 20mg을 분석 저울로 칭량하고, DMSO를 적당량 첨가하여 충분히 혼합 및 용해시켰다. 그 후, 상기에서 얻은 용액을 피펫으로 투석백에 적가하고, 증류수를 매시간 1회 교체하였다. 3시간 후 증류수를 2시간마다 교체하고 DMSO가 완전히 투석될 때까지 48시간 동안 매번 400~800mL의 증류수를 추가했습니다. 그 후, 위의 용액을 피펫으로 메스플라스크에 옮겨 일정한 부피를 얻은 다음 초음파로 2분간 처리하였다. 0.45μm 필터 멤브레인을 통한 여과로 ICG 라벨이 부착된 DZP-CHP 나노입자(ICG-DZP-CHP)가 생성되었으며, 이를 별도로 포장하여 향후 사용을 위해 4°C의 냉장고에 보관했습니다.
유화 형광 도네페질 CHP 나노입자의 제조
10mL 비이커에 ICG-DZP-CHP 적당량을 넣고, 1‰(v/v) 폴리소르베이트 80(PS) 유화제를 첨가하였다. 비이커를 1시간 동안 보관한 다음 100W에서 2분 동안 초음파 처리를 위해 EP 튜브로 옮겼습니다. 균일한 나노 용액이 얻어질 때까지 위의 조작을 3회 반복했습니다. 마지막으로, 여과되고 ICG로 표지된 유화 도네페질 약물이 로딩된 나노입자를 얻었다(PS-ICG-DZP-CHP).
나노입자에 대한 APOE의 결합을 확인하기 위한 MST 실험
모든 MST 실험은 Monolith NT.115 시스템(201810-BR-N024)에서 수행되었습니다. 모든 용액은 탈이온수와 분석용 시약으로 준비되었습니다. 버퍼를 준비하고 실온에서 보관했습니다. 단백질 샘플은 사용할 때까지 얼음에 보관되었습니다[33]. PS-ICG-CHP 나노입자(55.6μM)를 탈이온수로 40nM으로 희석하고 형광을 위해 ICG를 로드했습니다. APOE 용액(30μl, 55.6μM)을 준비하고 16개의 모세관을 1에서 16으로 표시했습니다. 먼저, 튜브 1에 APOE 20μl, 튜브 2~16에 10μl를 가했습니다. 그런 다음 튜브 1에서 튜브 2로 용액 10μL를 옮기고 완전히 혼합했습니다. 그 후, 10μl의 용액을 튜브 2에서 제거하고 튜브 3으로 옮겼습니다. 이 작업을 튜브 16에서 최종적으로 10μL의 용액이 제거될 때까지 반복하여 각 튜브의 용액이 동일한 부피가 되도록 보장했습니다. 10 마이크로리터의 희석된 나노입자를 각 튜브에 첨가하고 완전히 혼합하여 측정을 시작했습니다. MST 테스트 데이터는 NT-analysis 소프트웨어로 분석되었고 KD 피팅은 소프트웨어 지침에 따라 질량 작용의 법칙에 따라 수행되었습니다.
뇌 표적 관찰을 위한 생체 내 형광 이미징 기술
체중이 각각 약 18~22g인 건강한 검은색 마우스를 선택하고 PS-ICG-DZP-CHP 그룹과 ICG-DZP-CHP 그룹의 2개 그룹으로 무작위로 나누었습니다. 모든 마우스에 꼬리 정맥을 통해 200μg/ml의 위 그룹 약물 200μl를 주입하고 0.5시간 후 1% 펜토바르비탈 나트륨(50mg/kg)으로 마우스를 마취시켰다. 그 후 모든 마우스를 여기 파장 765nm-815nm 및 흡수 파장 815nm-845nm로 설정한 이미징 매개변수를 사용하여 라이브 이미저의 촬영 영역에 배치하여 전체 동물의 형광 이미지를 얻었습니다. . 이미징 후 모든 마우스를 해부하고 신장, 심장, 비장, 폐, 간 및 뇌를 제거하여 형광 이미지를 얻었다. 이미징 매개변수는 위에서 설명한 것과 일치했습니다.
나노입자의 조직 분포에 관한 연구
쥐 그룹화 및 샘플링
45마리의 C57BL/6 마우스를 무작위로 15개 그룹으로 나누었습니다:5개 그룹에는 유리 도네페질(유리 그룹), 5개 그룹에는 도네페질 나노입자(나노 그룹), 그리고 다른 5개에는 PS-변형 도네페질 나노입자(PS 그룹)가 주입되었습니다. 정맥 꼬리를 통해 0.25mg/kg. 그런 다음, 주사 후 1시간, 3시간 및 6시간에 혈액을 채취했습니다. 그 후 모든 동물을 희생시키고 심장, 뇌, 간, 신장 조직을 채취하여 파쇄하였다. 그 다음, 상기 조직 0.2g을 정밀히 달아 0.9% NaCl 용액 약 1ml에 가하고 균질화기(65Hz, 150초)로 균질화하였다. 100마이크로리터의 조직 균질액을 0.7mL의 메탄올이 포함된 1.5mL EP 튜브에 정확하게 끌어넣고 볼텍싱하여 단백질 침전을 위해 30초 동안 혼합한 다음 12,000r·min
-1
에서 원심분리합니다. 10분 동안 마지막으로 100μL의 상층액을 분석을 위해 주입 병에 옮겼습니다.
결정 방법
처음에는 HPLC를 이용하여 검사를 하였지만 감도가 충분히 높지 못하였다. 따라서 후속 LC-MS 실험이 수행되었으며, 이는 강한 특이성과 약물 결정을 방해하는 내인성 물질이 없음을 보여주었습니다. LC-MS 프로토콜은 생물학적 시료 측정 지침을 준수했습니다. 크로마토그래피 조건은 다음과 같았다:이동상 A, 물(0.1% 포름산 함유); 이동상 B, 메탄올(0.1% 포름산 함유); 등용매 용리:A30%-B70%; 유속, 0.3 mL· min
−1
; 컬럼 온도, 35°C; 주입 부피, 10 μL. 충돌 조건은 다음과 같습니다. 전자분무 이온화 소스(ESI) 온도, 150°C; 황폐화 가스 유량, 550L·h
−1
; 황폐화 가스 온도, 500°C. 양이온 검출을 위한 조건은 다음과 같았다:모세관 전압, 3kV; 콘 전압, 30V; 스캐닝 모드, 다중 반응 모니터링(MRM).
세포 실험
세포 배양 및 계대
PC12 및 SH-SY5Y 세포를 10%(v/v) 열불활성화 소태아혈청(FBS) 및 1%(v/v) 페니실린 및 스트렙토마이신이 보충된 고당 DMEM에서 배양한 다음 5를 포함하는 인큐베이터에 보관했습니다. % CO2 37°C에서 세포는 80% 합류에 도달하자마자 다양한 실험에 사용되거나 계대되었습니다. 실험 전에 PC12 및 SH-SY5Y 세포를 실험 규모에 따라 필요한 세포 밀도로 콜라겐 유형 I 사전 코팅 플레이트에 접종했습니다.
세포 동결보존
현미경하에서 대수기로 성장하는 것이 관찰되면, PC12 및 SH-SY5Y 세포를 동결시키고 PBS로 2회 세척하고, 트립신 처리하여 세포 현탁액을 형성하고 원심분리 수집을 위해 멸균 원심분리 튜브에 넣었다(1000 r x min
- 1
, 3분). 그런 다음 세포 동결보존 용액을 첨가하고 세포 이름과 날짜가 표시된 튜브에 세포를 보관했습니다. 세포를 4°C에서 1시간, − 20°C에서 2시간, − 80°C(동결)의 냉장고에 하룻밤 두어 최종적으로 액체 질소 탱크로 옮겼습니다.
세포 생존율 검출을 위한 MTT 방법
세포 생존율은 MTT 감소 분석을 사용하여 측정되었습니다. 간단히 말해서, PC12 및 SH-SY5Y 세포를 1 × 10
4
의 밀도로 96웰 플레이트(I형 콜라겐으로 미리 코팅)에 접종했습니다. 세포가 각 웰에 부착될 수 있도록 세포/mL. 24시간 동안 배양한 후 세포를 다양한 농도의 도네페질 CHP 나노용액 또는 유리 도네페질 용액과 함께 2시간 동안 사전 배양했습니다. 그 후, Aβ25-35(최종 농도 20μM)를 각 웰에 첨가했습니다. 처리된 96웰 플레이트를 37°C에서 24시간 동안 인큐베이션했습니다. 그 후, MTT(50μL, 5mg/mL)를 첨가하고 처리된 세포와 함께 37°C에서 4시간 동안 인큐베이션했습니다. 마지막으로 배지를 조심스럽게 제거하고 포르마잔 결정을 DMSO 150μL에 녹였습니다. 마이크로플레이트 판독기를 사용하여 490nm에서 흡광도를 얻었습니다. 세포 생존율은 처리군의 살아있는 세포의 백분율과 처리되지 않은 대조군의 살아있는 세포의 백분율로 표시됩니다.
세포 상청액의 LDH 활성 측정
PC12 및 SH-SY5Y 세포를 2 × 10
5
의 밀도로 96웰 배양 플레이트(유형 I 콜라겐으로 미리 코팅됨)에 접종했습니다. 및 3 × 10
5
세포/mL, 각각. 24시간 동안 배양한 후 세포를 다양한 농도의 도네페질 CHP 나노용액 또는 유리 도네페질 용액과 함께 2시간 동안 사전 배양했습니다. 그 후, Aβ25-35(최종 농도 20μM)를 각 웰에 첨가했습니다. LDH 활성은 키트에 제공된 지침에 따라 측정되었습니다. 간단히 말해서, 배지와 함께 배양된 세포를 수집한 다음 3500rpm에서 원심분리했습니다. 상등액(50 μL)을 동일한 부피의 반응물과 혼합하여 LDH 반응을 개시하였다. 마이크로플레이트 리더를 사용하여 450nm에서 흡광도를 구하고 LDH 활성을 계산했습니다.
아폽토시스 형태를 관찰하기 위한 AO/EB 염색 방법
AO/EB 형광 염료를 사용하여 세포 사멸 세포의 특성을 평가했습니다. PC12 및 SH-SY5Y 세포를 3 × 10
5
의 밀도로 검은색 12웰 배양 플레이트(I형 콜라겐으로 미리 코팅)에 접종했습니다. 및 4 × 10
5
세포/웰, 각각. 24시간 동안 배양한 후 세포를 다양한 농도의 도네페질 CHP 나노용액 또는 유리 도네페질 용액과 함께 2시간 동안 사전 배양했습니다. 그 후, Aβ25-35(최종 농도 20μM)를 각 웰에 첨가했습니다. 치료 후 키트에 따라 수술을 진행하였다. 실험 내내 빛을 피했습니다. 마지막으로 세포의 형태를 관찰하였다.
미토콘드리아 막 전위 검출을 위한 로다민 123 염색 방법
MMP는 로다민 123(Rh123) 형광 염료를 사용하여 측정되었으며, 이는 매우 부정적인 특성으로 인해 미토콘드리아에 우선적으로 분포하는 세포 투과성 양이온 염료입니다. PC12 및 SH-SY5Y 세포를 2 × 10
5
의 밀도로 검은색 24웰 배양 플레이트(유형 I 콜라겐으로 미리 코팅)에 접종했습니다. 및 3 × 10
5
세포/웰, 각각. 24시간 동안 배양한 후 세포를 다양한 농도의 도네페질 CHP 나노용액 또는 유리 도네페질 용액과 함께 2시간 동안 사전 배양했습니다. 그 후, Aβ25-35(최종 농도 20μM)를 각 웰에 첨가했습니다. 처리 후, 세포를 PBS로 세척하고 10μg/mL 로다민 123과 함께 37°C의 암실에서 30분 동안 인큐베이션했습니다. 배양 후 세포를 PBS로 3회 세척하고 형광 플레이트 리더를 사용하여 488 nm 및 510 nm에서 형광 강도를 측정했습니다.
통계 처리 및 데이터 분석
모든 실험은 3회 반복하였으며, 그 결과를 평균 ± 표준편차로 나타내었다. GraphPad Prism 통계 소프트웨어가 사용되었으며 일원 ANOVA, Student's t 테스트 및 기타 방법을 사용하여 통계적 분석을 수행했습니다. 피 <0.05는 차이가 통계적으로 유의함을 나타냅니다.
<섹션 데이터-제목="결과">
결과
나노입자의 특성
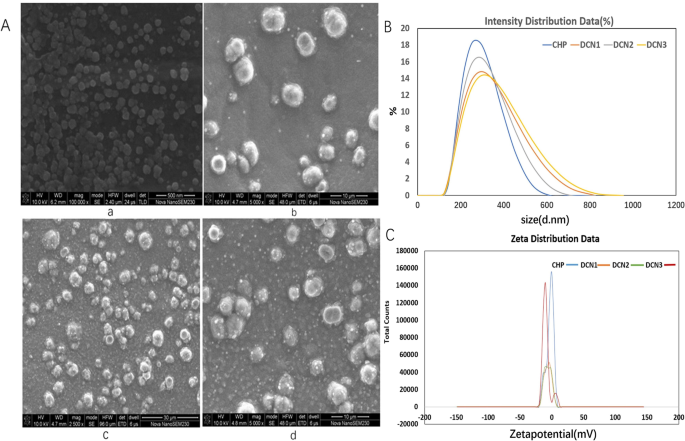
CHP는 DZP를 탑재할 수 있는 소수성 코어가 있는 나노입자를 형성하기 위해 자체 응집합니다. 약물 대 나노물질 비율이 1:2, 1:5 및 1:10인 DZP가 장착된 CHP 나노입자(DCN)는 DCN1, DCN2 및 DCN3으로 명명되었습니다. 주사전자현미경 결과에 따르면, CHP 나노입자는 구형 구조를 나타내었고, DZP 로딩 후 DCN도 그림 1과 같이 구형이었다. CHP 나노입자의 평균 크기와 제타 전위는 257 ± 3.05 nm 및 -2.81이었다. 각각 ± 0.27mV입니다. DZP 로딩 후, 평균 크기는 각각 273 ± 3.72, 260.7 ± 1.76 및 266.8 ± 4.56 nm이었고, DCNm, DCNm, N 에 대해 1, N , 0.40, − . , 표 1에 나타난 바와 같다. 약물의 포집율은 42.00 ± 5.65%, 86.54 ± 1.31% 및 59.71 ± ,.3,.3,.3,.2,12.02 .90± 12.02 .9± 각각.
<그림>
주사 전자 현미경 이미지(a , a-CHP, b-DCN1, c-DCN2, d-DCN3), 크기 분포(b a-CHP, b-DCN1, c-DCN2, d-DCN3) 및 제타 전위(c a-CHP, b-DCN1, c-DCN2, d-DCN3) 나노입자
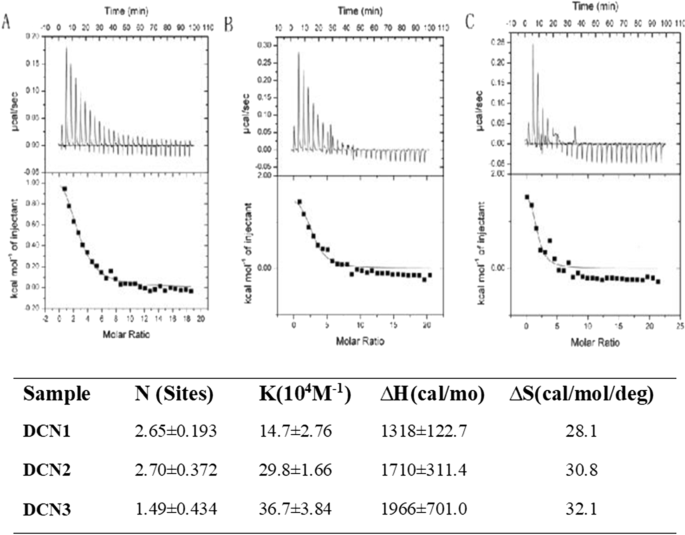
ITC 측정
DCN의 경우 전체 반응 과정에서 반응이 주로 상향 피크를 보였고(그림 2), 상향 피크가 열 방출 반응을 나타내기 때문에 흡열 반응이었다. 따라서 PS는 DCN 표면에 자발적으로 흡착될 수 있습니다. PS 친화도는 (14.7 ± 2.76) × 10
4
M
−1
, (29.8 ± 1.66) × 10
4
M
−1
그리고 (36.7 ± 3.84) × 10
4
M
−1
, 그리고 PS 커버리지의 정도는 DCN1, DCN2 및 DCN3에 대해 각각 2.65 ± 0.193, 2.70 ± 0.372 및 1.49 ± 0.434였습니다. 이 결과는 PS가 DCN 표면에 높은 친화력으로 흡착되고 DCN2에 더 많은 커버리지를 가짐을 나타냅니다. ∆H> 0 및 ∆S> 0은 세 입자가 주로 PS와의 소수성 상호작용을 통해 결합되었음을 나타냅니다.
<사진>
a로의 PS(0.9mM) 적정을 위한 등온 열량계 데이터 DCN1, b DCN2 및 c 25°C에서 DCN3(0.02mM) 솔루션 NP 용액으로 적정한 후 PS가 나노입자(NP)와 결합하는 범위, 친화도(KA), 엔탈피 및 엔트로피 변화
세 가지 나노입자 유형의 특성
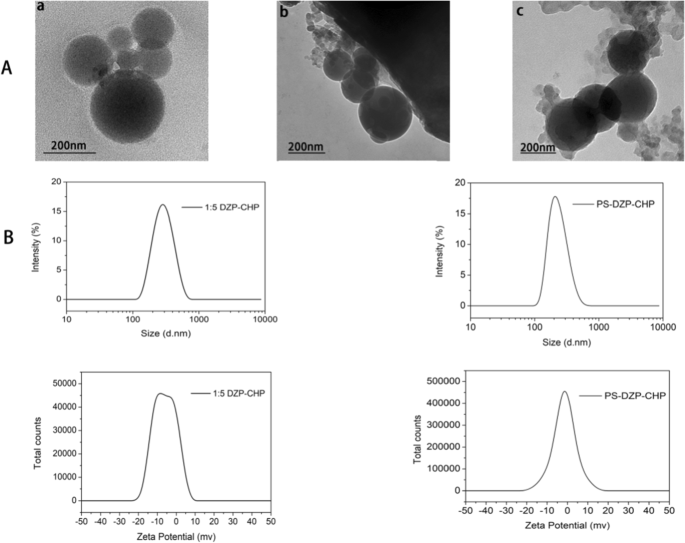
투석으로 제조된 CHP NP와 DCN은 균일한 구형을 보였다(그림 3a). 위의 연구에 따라 우리는 약물 대 나노 물질의 비율이 1:5인 DZP-CHP 나노 입자를 다음 실험의 대상으로 선택했습니다. DZP-CHP 나노입자는 260.7 ± 1.76nm의 비교적 균일한 입자크기를 가지며, 분산지수는 0.196 ± 0.019였다. 입자 크기는 약물 로딩 후 비교적 안정적으로 유지되었지만 PS 흡착 후 335.2 ± 5.46 nm로 급격히 증가했습니다. DZP-CHP 나노입자의 제타 전위는 -0.66 ± 0.04mV였으며, 폴리소르베이트 80으로 코팅한 후 제타 전위는 -2.22 ± 0.86mV로 떨어졌습니다(그림 3b).
<그림>
다른 나노 입자의 특성화. 아 a CHP NP의 투과 전자 현미경 사진, b DZP-CHP NP의 투과 전자 현미경 사진, c PS-DZP-CHP NP의 투과 전자 현미경 사진. B:CHP-DZP NP(공급비 1:5) 및 폴리소르베이트 80(공급비 1:5)으로 변형된 DZP-CHP NP의 입자 크기 다이어그램 및 제타 전위 다이어그램
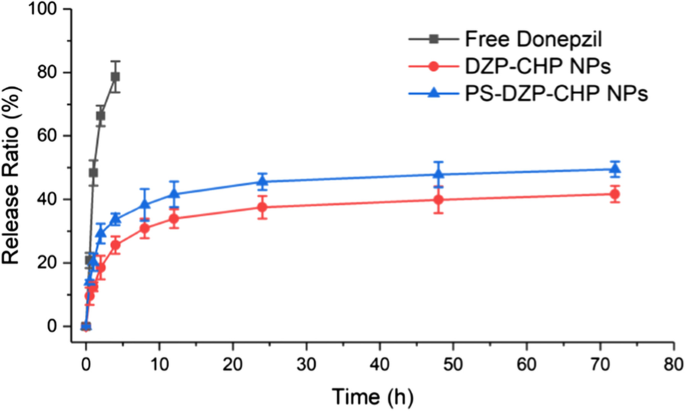
나노입자의 체외 약물 방출
결과는 유리 도네페질과 비교하여 DZP-CHP NP 및 PS-DZP-CHP NP가 72시간 동안 DZP를 방출하고 분명한 제어 방출 효과가 있음을 보여주었습니다. 약물이 담지된 나노입자의 빠른 빠른 방출 속도는 약물 분자의 빠른 용해와 방출로 인한 것일 수 있으며, 그 다음에는 약물 농도의 감소로 인해 약물 농도의 저하가 발생할 수 있으며, 이는 용해 및 확산에 의해서만 영향을 받을 수 있습니다. 그런 다음 폴리소르베이트 80으로 코팅 및 코팅되지 않은 DZP-CHP NP의 시험관 내 약물 방출을 연구했습니다. PS-DZP-CHP NPs의 느린 방출은 폴리소르베이트 80이 작은 소수성 분자 약물에 강하게 흡착되기 때문일 수 있습니다(그림 4).
<그림>
DZP, DZP-CHP NP(1:5) 및 PS-DZP-CHP NP에 대한 시험관 내 약물 방출 곡선
나노입자 뇌 표적 효과
라이브 형광 이미징 기술을 사용한 뇌 표적화 관찰
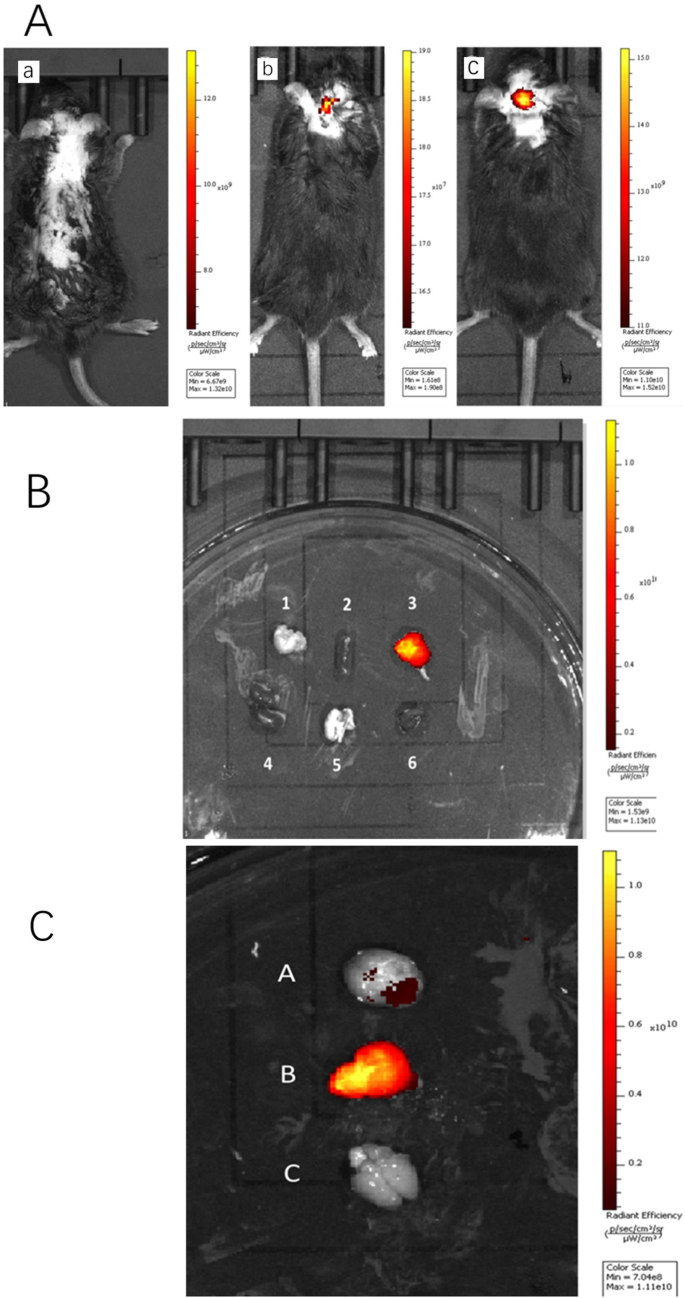
유리 ICG를 주사한 쥐의 뇌는 형광을 나타내지 않았지만 PS로 유화된 나노입자를 주사한 뇌는 비유화 나노입자를 주사한 뇌보다 뇌에서 더 강한 형광을 나타냈다. . 5a). 이를 확인하기 위해 ICG-DZP-CHP 및 PS-ICG-DZP-CHP 용액을 정맥 주사한 후 30분 후에 마우스를 해부하고 연구에 필요한 모든 장기를 제거한 다음 형광 이미징을 수행했습니다. PS-ICG-DZP-CHP 나노 입자는 뇌에서 강한 형광을 나타내었지만 다른 기관에서는 관찰되지 않았습니다(그림 5b). 이미지는 PS로 변형된 나노입자가 뇌 조직에서 가장 강한 형광을 나타내는 반면 변형되지 않은 나노입자는 약한 형광을 나타냄을 보여주었다. 유리 ICG를 주입한 마우스의 뇌 조직에서는 형광이 관찰되지 않았습니다(그림 5c).
<그림>
꼬리 정맥을 통해 다양한 용액을 주입한 후의 생체 내 형광 이미지. 아 200μg/ml DZP-CHP 또는 PS-DZP-CHP 나노입자가 ICG를 얼룩으로 로드한 꼬리 정맥을 통해 주입된 전체 동물의 형광 이미지. 무료 ICG 용액(형광 강도 × 10
9
), b ICG-DZP-CHP 나노입자(형광 강도 × 10
7
), ㄷ PS에 의해 변형된 ICG-DZP-CHP 나노입자(형광 강도 × 109). ㄴ Fluorescence images of various organs after injection of DZP-CHP nanoparticles modified by PS via the tail vein. ㄷ Fluorescence images of the brain after dissection. d Brain of mice injected with ICG-PS-DZP-CHP nanoparticles, e brain of mice injected with ICG-DZP-CHP nanoparticles modified with PS, f brain of mice injected with free ICG
Tissue Distribution of Nanoparticles in Mice
Donepezil was distributed in various tissues and mainly in the brain after injection of PS-DZP-CHP nanoparticles. Because it is metabolized through the kidney, the concentration of donepezil is very high in the kidney at certain times (Fig. 6a). In the brain, the concentration of free donepezil reached a peak in a very short time and then decreased rapidly. However, the concentration of donepezil nanoparticles reached a peak much more slowly and then decreased, especially nanoparticles modified with PS, which indicates a sustained-release effect with a delayed peak and prolonged retention time. Obviously, the nanoparticles improved the bioavailability of the drug (Fig. 6b).
아 The concentration of donepezil in the brain, heart, liver and kidney at different times. ㄴ The concentrations of free DZP, DZP-CHP nanosolution and DZP-CHP nanosolution modified with PS in brain tissue at different times
MST Results
MST results showed that the thermal surge changed regularly with increasing ligand concentration, and the KD value was 3.63 μM, indicating that the ligand effectively binds to the target protein, which verifies that PS can bind to Apo E and is relatively stable. After surface modification with PS, CHP nanoparticles can promote adsorption of Apo E, theoretically confirming that the nanoparticles we designed can specifically target brain tissue because the nanoparticles adsorbed Apo E, which may mediate passage through the blood–brain barrier (Fig. 7).
아 Raw MST data. The fluorescently labeled molecules were observed for 5 s. At this time, the infrared laser was turned on, and a small part of the capillary tube was heated to 2–5 °C. The molecules migrated along a thermal gradient, resulting in changes in fluorescence intensity. When the infrared laser is turned off, the molecules diffuse along the concentration gradient. ㄴ The binding curve is generated by the difference between the initial fluorescence intensity and the intensity in the presence of heat, and the curve conforms to the standard 1:1 binding model
Establishment of a Nerve Injury Model Induced by Aβ25–35
MTT tests were used to detect the effect of different concentrations of Aβ25-35 on the activity of PC12 and SH-SY5Y cells [Fig. 8a (i), Fig. 8b (i)], and the results showed that with increasing Aβ25–35 concentration, PC12 and SH-SY5Y cell proliferation activity gradually decreased compared with that in the normal control group. When PC12 and SH-SY5Y cells were treated with 20 μM Aβ25–35 , PC12 cell activity decreased to 49.5 ± 3.3% that observed in the control group (P < 0.01), and SH-SY5Y cell activity decreased to 49.7 ± 0.8% (P < 0.01). An LDH kit was used to detect the effect of different concentrations of Aβ25–35 on LDH activity in both cell supernatants. Colorimetric tests showed that activity in both supernatants increased gradually with increasing Aβ25–35 concentration. After treatment of the cells with 20 μM Aβ25–35 , PC12 cell LDH release increased to 359.3 ± 18.3% that in the control group (P < 0.01), and SH-SY5Y cell LDH release increased to 360.0 ± 18.2% (P < 0.01). Rhodamine 123 staining was used to detect the effect of different concentrations of Aβ25– 35 on the mitochondrial membrane potential in both cell lines [Fig. 8a (ii), b (ii)]. The test showed that the mitochondrial membrane potential in both cell lines decreased gradually with increasing Aβ25– 35 concentration (5, 10, 20, 40 μmol/L). Treatment of the cells with 20 μM Aβ25–35 decreased the PC12 cell mitochondrial membrane potential to 51.3 ± 1.6% that in the control group (P < 0.01); for SH-SY5Y cells, the MMP decreased to 47.9 ± 1.7% that in the control group (P < 0.01).
Effects of different concentrations of Aβ25–35 on injury to PC12 and SH-SY5Y cells. 아 , b The effect of different concentrations of Aβ25–35 on the cell survival rate, LDH activity and cell mitochondrial membrane potential in PC12 cells and SH-SY5Y cells (*P < 0.05, ** P < 0.01 vs control group). ㄷ The effect of different concentrations of Aβ25– 35 on damage to the morphology of PC12 cells:a control group, b Aβ25– 35 (5 μM) injury group, c Aβ25– 35 (10 μM) injury group, d Aβ25– 35 (20 μM) injury group, e Aβ25– 35 (40 μM) injury group. D:the effect of different concentrations of Aβ25– 35 on injury to the morphology of SH-SY5Y cells:f—control group, g Aβ25– 35 (5 μM) injury group, h—Aβ25– 35 (10 μM) injury group, i—Aβ25– 35 (20 μM) injury group, j—Aβ25– 35 (40 μM) injury group
Inverted fluorescence microscopy was used to observe morphological changes of PC12 and SH-SY5Y cells injured by different concentrations of Aβ25– 35 (Fig. 8c, d). The PC12 and SH-SY5Y cells in the control group had higher density, fusiform shapes, fuller cell bodies and longer protrusions. As the concentration of Aβ25– 35 increased (5, 10, 20, 40 μmol/L), the number of cells in both cell lines gradually decreased, the cell bodies shrank slightly, and the protrusions began to shrink sharply. When the concentration of Aβ25– 35 was increased to 40 μmol/L, the protrusions broke significantly, most of the cells contracted sharply, their shape became irregular, and some cells detached and became suspended in the solution.
Therefore, we treated PC12 and SH-SY5Y cells with 20 μM Aβ25-35 for 24 h to establish a nerve injury model.
Neuroprotective Effect of Drug-Loaded Nanoparticles (DZP-CHP)
MTT assays were used to detect the activity of different concentrations of DZP and DZP-CHP (2.5 μM, 5 μM, 10 μM) in PC12 and SH-SY5Y cells [Fig. 9a (i), b(i)]. Tests showed that treatment of PC12 cells with 20 μM Aβ25-35 alone resulted in a significant reduction in cell viability to 48.4 ± 2.8% that in the control group (P < 0.01). However, after pretreatment with DZP and DZP-CHP (2.5 μM, 5 μM, 10 μM) solutions, the viability of PC12 cells increased significantly. The viability of PC12 cells in the DZP-CHP group was higher than that in the DZP group (P <0.05). Similarly, treatment of SH-SY5Y cells with 20 μM Aβ25– 35 alone resulted in a significant reduction in cell viability to 48.5 ± 4.0% that in the control group (P < 0.01), while after pretreatment with DZP and DZP-CHP solution (2.5 μM, 5 μM, 10 μM, respectively), the viability of SH-SY5Y cells increased significantly. The viability of SH-SY5Y cells in the DZP-CHP group was higher than that in the DZP group (P <0.05).
Effect of DZP-CHP on Aβ25-35-injured PC12 and SH-SY5Y cells. 아 and b The effects of DZP-CHP on the cell survival rate, LDH activity, and mitochondrial membrane potential of PC12 and SH-SY5Y cells injured by Aβ25-35 (#P < 0.05, ## P < 0.01 vs Aβ25-35 group; ▲ P < 0.05 vs DZP group). ㄷ The effect of DZP-CHP on the apoptosis morphology of PC12 cells injured by Aβ25-35; a—control group, b—Aβ25-35 injury group, c—DZP (5 µM), d—DZP -CHP (5 µM), e—DZP (10 µM), f—DZP-CHP (10 µM). D:the effect of DZP-CHP on the apoptosis morphology of SH-SY5Y cells injured by Aβ25-35; g—control group, h—Aβ25-35 injury group, i—DZP (5 µM), j—DZP-CHP (5 µM), k—DZP (10 µM), l—DZP-CHP (10 µM). E:red/green fluorescence ratio
LDH kits were used to detect the effects of different concentrations of DZP and DZP-CHP (5 μM and 10 μM) on the release of LDH from PC12 and SH-SY5Y cells into the culture medium [Fig. 9a (ii), b (ii)]. Colorimetric measurements showed that the release of LDH from PC12 cells that were exposed to 20 μM Aβ25-35 alone increased significantly by 355.1 ± 16.6% (P < 0.01). In the presence of DZP and DZP-CHP (5 μM and 10 μM), LDH release from PC12 cells dropped significantly. The effect in the DZP-CHP group was higher than that in the DZP group (P < 0.01). Similarly, the release of LDH from SH-SY5Y cells exposed to 20 μM Aβ25-35 increased significantly to 357.8 ± 12.5% (P < 0.01). However, after pretreatment with DZP and DZP-CHP (5 μM and 10 μM), LDH release from SH-SY5Y cells decreased significantly. The effect in the DZP-CHP group was higher than that in the DZP group (P <0.05).
According to previous reports, depolarization of the MMP leads to loss of Rh123 from mitochondria, which in turn leads to a decline in intracellular fluorescence. Therefore, to characterize changes in the mitochondrial membrane potential in PC12 and SH-SY5Y cells treated with Aβ25-35, DZP, and DZP-CHP (5 μM, 10 μM), rhodamine 123 was used for detection [Fig. 9a (iii), b (iii)]. The results showed that the fluorescence intensity of rhodamine 123 decreased significantly to 44.3 ± 3.8% (P < 0.01) after incubation of PC12 cells with 20 μM Aβ25-35 for 24 h. However, pretreatment with DZP and DZP-CHP (5 μM, 10 μM) solutions resulted in a significant increase in fluorescence intensity in a dose-dependent manner, and the effect in the DZP-CHP group was higher than that in the DZP group (P <0.05). Similarly, after treatment of SH-SY5Y cells with 20 μM Aβ25-35 for 24 h, the fluorescence intensity of rhodamine 123 significantly decreased to 42.5 ± 4.6% (P < 0.01). However, after pretreatment with DZP and DZP-CHP (5 μM, 10 μM), the fluorescence intensity increased significantly, and the effect in the DZP-CHP group was higher than that in the DZP group.
An AO-EB double staining kit was used to detect morphological changes of PC12 and SH-SY5Y cells treated with different concentrations of DZP and DZP-CHP (5 μM and 10 μM) (Fig. 9c, d). After AO-EB double staining, the nuclei of living cells presented green fluorescence under a fluorescence, and the fluorescence of apoptotic cells was orange-red; the higher the degree of apoptosis is, the brighter the fluorescence. Compared with the untreated control group, PC12 and SH-SY5Y cells treated with Aβ25-35 alone showed typical apoptotic characteristics, such as highly condensed and broken nuclei and obvious cell injury. However, pretreatment with DZP and DZP-CHP solution (5 μM and 10 μM) significantly inhibited cell damage and improved cell morphology.
Discussion
Although nanoparticles have been shown to be an effective delivery medium for nervous system diseases, the complexity of their structure and performance makes it challenging to detect and evaluate their physical–chemical properties and biological safety [34,35,36]. Therefore, after nanodrugs are designed, experiments to verify the safety and effectiveness of the nanomaterials at the cell and animal levels are extremely necessary. In the early laboratory stage, PS-DZP-CHP nanoparticles were successfully synthesized, and the optimal dosing ratio of the drug to CHP was set at 1:5. Due to the stable adsorption of PS to apolipoproteins ApoB and ApoE, brain targeting of nanoparticles can be achieved by permeation through the BBB [37]. The expected goal is that nanoparticles begin to decompose after reaching the brain; DZP is released and increases the concentration of cholinesterase; CHP reduces Aβ protein deposition and improves the brain environment; and administration frequency decreases because of the sustained release from nanoparticles [38,39,40,41].
Therefore, this study conducted an in vitro drug release test to assess the sustained release effect of nanoparticles. Compared with free DZP, nanoparticles achieved local sustained release in the brain, and PS can adsorb plasma proteins and reduce the loss of nanoparticles and prolong the release time to achieve a long cycle. After injection of ICG-labeled DZP-CHP and PS-DZP-CHP nanoparticles into rats, emulsified nanoparticles showed stronger fluorescence in the brain than those not emulsified with Tween 80. After organ biopsy, fluorescence imaging of various organs revealed that only the brain presented strong fluorescence, while other organs did not, indicating that nanoparticles did not release the drug until they reached the brain, which met the expected goal. In addition, nanoparticles modified with polysorbate 80 adsorbed ApoE to the surface and simulated low-density lipoprotein to bind to lipoprotein receptors on the surface of endothelial cells and enter the brain through LDLR induction [42, 43]. Moreover, the mechanisms by which nanoparticles regulate the tight junctions between endothelial cells and the inhibition of P-glycoprotein may produce a synergistic effect on their transcellular transport into the brain parenchyma [44]. However, since polysorbate 80 is prone to produce toxic substances, which cause untoward reactions, such as hypotension, dyspnea and shock, the dosage should be strictly controlled [45, 46]. Although a large number of nanodrugs have been developed for treatment of CNS diseases, most have shown poor effects [4]. In this study, we built a brain model of AD patients to evaluate the protective effect of nanoparticles on the brain. Because the toxicity of the Aβ protein to nerve cells varies with the concentration, it is better to screen an appropriate concentration to better simulate the brain environment of AD patients. The model was treated with DZP and DZP-CHP nanosolutions in advance to investigate the protective effect of DZP-CHP against PC12 and SH-SY5Y cell damage induced by Aβ25-35. The results showed that both solutions improved the cell proliferation activity caused by Aβ25-35, LDH release declined, and the mitochondrial potential rose. The inhibitory effect of the DZP-CHP nanosolution on cell damage induced by Aβ25-35 was significantly better than that of free DZP solution, and a concentration of 10 M DZP-CHP nanosolution proved to be the best, which may be due to the optimal drug concentration approach.
Basically, this study deduced the activity process of nanoparticles in vivo , verifying that PS-DZP-CHP nanoparticles have strong brain targeting, a good sustained release effect and a good AD therapeutic effect, thus demonstrating that they are clinically promising nanodrugs. The difficulty of treating the brain with medication has always been a major problem for researchers. The BBB leads to difficulty in curing CNS diseases, such as Parkinson's disease, brain tumors and brain stroke [47]. PS-DZP-CHP nanoparticles can effectively pass the BBB without destroying it, and DZP can be replaced as a model drug. Loading other drugs with high fat solubility and poor dissolution in vivo into the hydrophobic center of the NPs can not only increase brain targeting but also improve the water solubility of the drug. In addition, the design of CHP nanoparticle solutions is simple, and the drug dosage of the hydrophobic center can be flexibly controlled to ensure the best therapeutic effect while minimizing cytotoxicity [48, 49]. In future work, we will conduct in vivo research on DZP-CHP nanoparticles, such as evaluation of drug metabolism and side effects. These studies supplied a new strategy for brain drug delivery, and it is advantageous to clinical application of nanodrug with AD treatment.
Conclusion
DZP-CHP nanoparticles showed an optimal drug to nanomaterials dosing ratio of 1:5, which led to higher PS coverage and drug loading. DZP-CHP nanoparticles with PS adsorption exhibited slow release and significant brain targeting. Nanoparticle surface modification with PS can promote adsorption of Apo E and thus is vital for brain targeting. DZP-CHP nanoparticles had a protective effect on neurotoxicity and were superior to free donepezil.