저에너지 스핀트로닉스에서 시급히 필요한 것은 액체-질소 온도(77K) 이상의 퀴리 온도와 상당한 자기 이방성을 갖는 2차원(2D) 강자성체입니다. 우리는 Mn3을(를) 공부했습니다. Br8 MnBr2에서 인구의 1/4에서 Mn 공석을 유도하여 얻은 단층 단층. 이러한 결함 구성은 Mn-d
5
의 조정 구조를 변경하도록 설계되었습니다. 상당한 자기 이방성 에너지(MAE)로 강자성을 달성합니다. 우리의 계산에 따르면 Mn3 Br8 단층은 130K의 큐리 온도, 공식 단위당 − 2.33meV의 큰 MAE, 13/3μB의 원자 자기 모멘트를 갖는 강자성(FM) 반금속입니다. Mn 원자의 경우. 또한, Mn3 Br8 단층은 5% 압축 변형에서 퀴리 온도가 160K인 작은 이축 변형에서 FM을 유지합니다. 또한 이축 변형과 캐리어 도핑 모두 MAE를 증가시키며 이는 주로 MCE(자기 결정 이방성 에너지)에 의해 기여합니다. 우리가 설계한 MnBr2 결함 구조 단층은 2D 재료에서 큰 MAE로 강자성을 달성하는 간단하지만 효과적인 방법을 제공합니다.
소개
전자 스핀 및 관련 자기 모멘트를 이용하는 Spintronics는 전하 기반 장치에 비해 고유한 장점 때문에 지난 수십 년 동안 [1] 광범위한 관심을 받았습니다. 유한 온도에서 장거리 자기 정렬을 갖는 2차원(2D) 강자성[2, 3]의 최근 실현은 나노스케일 스핀트로닉스 및 관련 응용 분야에 매우 중요하며 2D 강자성[4,5 ,6,7,8,9].
원자 두께의 처음 두 개의 2D 강자성체, 즉 단층 CrI3 [2] 및 이중층 Cr2 Ge2 테6 [삼]. 불행히도 두 제품의 퀴리 온도는 모두 액체-질소 온도(77 K)보다 낮기 때문에 실제 적용이 제한됩니다. 퀴리 온도 외에 상당한 자기 이방성과 자기 모멘트도 실제 적용에 필수 불가결합니다. 큰 자기 이방성 에너지(MAE)는 열 변동에 대한 자기 정렬의 이점과 정보 비트당 입자 크기를 줄일 수 있는 가능성을 의미합니다. 작은 MAE는 강자성보다는 초상자성을 초래할 수 있습니다. 큰 자기 모멘트는 스핀트로닉스에 더 높은 감도, 더 높은 효율성 및 더 높은 밀도를 제공합니다. 무거운 원소는 강력한 SOC(spin-orbital coupling) 효과로 인해 큰 MAE를 유발할 가능성이 더 큽니다[10]. 무거운 원소로 구성된 일련의 2D FM 재료는 CrI3와 같은 큰 MAE를 갖는 것으로 예측되었습니다. [11], CrAs [12], CrSeI [13], CrSiTe3 [14], CrWI6 [15], FeBr2 및 FeI2 단층[16]. 또한, MXenes Mn2의 Mn 원자에 대한 국부 자기 모멘트 NF2 및 Mn2 N(OH)2 4.5μB 보고된 FM 2D 재료 중 가장 큰 Mn 원자당 [17]입니다.
CrI3 이후 단층이 성공적으로 합성되었고 전이 금속 할로겐화물은 많은 관심을 끌었다[18,19,20,21,22,23,24,25,26,27]. 이중층 MnF2에서 스핀 시백 효과가 관찰되었습니다. [20]; 몇 층의 CrI3 자기 터널링 접합(MTJ) [21]에 구현되었습니다. NiCl3 단층은 새로운 Dirac 스핀 갭리스 반도체(SGS)로 예측되었습니다[22]. 특히, MnBr2 단층은 첫 번째 원칙 계산을 기반으로 하는 평면에 수직 방향을 따라 0.25meV MAE를 갖는 반강자성입니다[16]. Mn
2+
이온은 d
5
에 있습니다. 자기 모멘트가 5μB인 높은 스핀 상태 [16, 26]. 이 결과는 MnBr2의 잠재력을 의미합니다. 자기 모멘트가 큰 단층 강자성체. 주요 문제는 Mn 이온 간의 AFM 커플링을 FM 커플링으로 변환하는 방법입니다.
LaMnO3에서 Mn 공석의 상당한 밀도가 실험적으로 관찰되었습니다. 박막[28], 고에너지 입자의 조사 또는 화학적 에칭[29]을 통해 합성 공정을 의도적으로 조절하여 결함의 농도를 제어할 수 있습니다. 이러한 맥락에서 우리는 Mn3 Br8 MnBr2에 단일 Mn 공석을 유도하여 단층 단층. 공석은 Mn 원자의 배위 구조를 변경하고 d
5
를 끊습니다. 이는 반강자성 커플링을 강자성 커플링으로 변환하고 무거운 Br 원자로 인해 큰 MAE를 가져올 수 있는 구성입니다. 예상대로 Mn3 Br8 단층은 FM이고 공식 단위당 − 2.33meV의 큰 MAE를 가지며, 각 Mn 원자의 자기 모멘트는 13/3μB입니다. . 유연한 기판을 구부림으로써 변형의 도입이 용이함을 고려[30,31,32,33], 탄성 기판을 길게 [33,34,35], 열팽창 불일치를 이용[33, 36] 등[33], 정전기 도핑을 통한 스핀 분극의 효과적인 제어 [37, 38], 우리는 또한 Mn3 Br8 이축 변형 및 캐리어 도핑 하에서의 단층. 우리의 결과는 Mn3 Br8 단층은 작은 이축 변형 하에서 퀴리 온도가 증가하면서 FM을 유지합니다. 또한 이축 변형과 캐리어 도핑 모두 MAE를 증가시킬 수 있습니다.
계산 방법
본 연구의 모든 계산은 Vienna ab-initio 시뮬레이션 패키지(VASP) [39]. 전자와 핵 사이의 상호 작용은 PAW(Projector Augmented Wave) 방법[40, 41]으로 설명되었으며 전자 교환-상관 상호 작용은 GGA(generalized gradient approximation) 내에서 Perdew-Burke-Ernzerhof(PBE) 기능으로 설명되었습니다. 방법[42]. 강력한 상관 상호 작용을 계산하기 위해 Hubbard U 항이 채택되었습니다[43]. Mn-incorporated 2D 재료 연구에 채택된 4 eV의 유효 현장 쿨롱 상호작용 매개변수(U)와 1 eV의 교환 에너지(J)가 Mn-d 전자에 사용되었습니다[44]. Brillouin 영역 통합은 Monkhorst-Pack 방식을 기반으로 하는 9 × 9 × 1 k-mesh를 채택하여 수행되었습니다[45]. 포논 스펙트럼은 VASP 패키지 내에서 구현되는 포노피 코드[46]를 사용하여 계산되었습니다. 인접한 층 사이의 상호 작용을 피하기 위해 단층 표면에 수직인 방향을 따라 20Å의 진공 공간을 추가했습니다. 평면파 기준 세트의 차단 에너지는 500eV로 설정되었습니다. 총에너지와 힘의 수렴기준은 1 × 10
–6
으로 설정하였다. 각각 eV 및 0.01eV/Å입니다.
결과 및 토론
MnBr의 분열 에너지, 바닥 상태 및 안정성2 단층
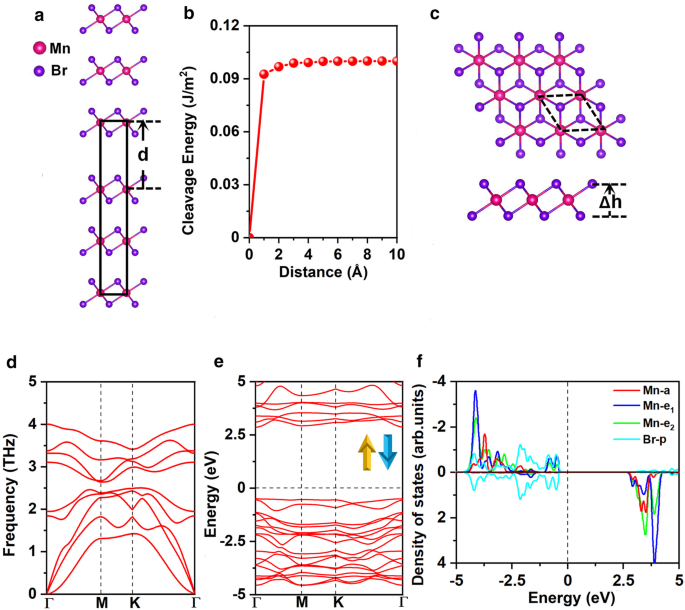
벌크 MnBr2의 최적화된 격자 상수 a =b =3.95 Å이며 이전 실험 결과와 일치합니다(a =ㄴ =3.87 Å) [25]. 우리는 먼저 MnBr2 각질 제거의 가능성을 조사했습니다. 벌크 MnBr2의 단층 . 그림 1a는 잘 알려져 있고 효과적이고 널리 승인된 분열 에너지 계산 방법을 나타냅니다[47,48,49]. 구체적으로, 벽개 에너지는 그림 1b와 같이 두 파단부 사이의 이격 거리 \(d\)에 대한 바닥 상태의 총 에너지 변화를 계산하여 구하였으며, 와 b의 격자 상수는 다음과 같다. 벌크 MnBr2의 평형 상태에서 값으로 고정 . 층간 장거리 vdW 상호 작용은 Grimme의 DFT-D2 방식으로 설명됩니다[50, 51]. 총 에너지는 이격 거리에 따라 증가하다가 그림 1b와 같이 천천히 수렴합니다. 계산된 절단 에너지는 0.10J/m
2
입니다. , 이는 흑연의 두 파단 부분 사이의 분열 에너지(0.35 J/m
2
)에 비해 작습니다. ) [52], MnBr2 획득 가능성 입증 미세 기계 박리 방법을 통한 단층.
<그림>
아 MnBr2의 벌크 모델 분열 에너지를 계산하는 데 사용되며 b 2개의 파단된 부분 사이의 분리 거리 \(d\)의 함수로서의 분열 에너지(평형 층간 거리는 0으로 설정됨). ㄷ 평면도 및 측면도, d 포논 스펙트럼, e 스핀 채널 및 f에 대한 전자 밴드 구조 MnBr2에 대한 Mn-d 궤도 및 Br-p 궤도의 예상 밀도(PDOS) 단층. Δh 두 할로겐화물 평면 사이의 수직 거리를 나타냅니다. 원시 세포는 검은색 점선으로 순환됩니다. 밴드 구조 및 DOS에 대한 페르미 레벨은 0eV로 설정됨
MnBr2 단층은 그림 1c와 같이 \(C_{{{3}v}}\) 대칭을 가지고 있습니다. 각 Mn 원자는 6개의 인접한 Br 원자로 둘러싸여 있어 팔면체 [MnBr6를 형성합니다. ]
4−
단위. 그림 2a 및 b에 표시된 것처럼 세 가지 가능한 자기 구성, 즉 비자성(NM), 강자성(FM) 및 반강자성(AFM) 상태가 고려됩니다. Mn 이온의 높은 스핀 상태와 낮은 스핀 상태가 모두 고려됩니다. 우리의 결과는 FM 상태의 Mn 이온이 d
1
로 낮은 스핀에 있음을 보여줍니다. 구성, AFM 상태의 Mn 이온은 d
5
로 높은 스핀 상태에 있습니다. 구성. MnBr2의 접지 상태 단층은 AFM 상태로, 공식 단위당 각각 3.91eV 및 0.72eV만큼 NM 및 FM 상태보다 더 안정적입니다(추가 파일 1:표 S1). MAE는 0.25meV이며, 양수 값은 용이한 자화 축이 면외 방향을 따라 있음을 나타내며 이전 결과와 일치합니다[16]. 최적화된 격자 상수는 a =ㄴ =3.95 Å, 벌크 MnBr2의 격자 상수와 동일 . Mn-Br 결합 길이는 2.73Å이고 두 할로겐화물 평면 사이의 수직 거리는 3.03Å입니다.
<그림>
a의 개략도 강자성 및 b MnBr2에 대한 반강자성 구성 단층
MnBr2의 안정성 형성 에너지, 포논 스펙트럼 및 탄성 상수를 계산하여 단층을 추가로 조사했습니다. 형성 에너지는 다음과 같이 계산됩니다.
여기서 \(E_{{{\text{MnBr}}_{{2}} }}\)는 MnBr2의 에너지를 나타냅니다. 단층, \(E_{{{\text{Mn}}}}\) 및 \(E_{{{\text{Br}}}}\)는 각각 벌크 구조에서 Mn 및 Br 원자의 에너지입니다. 계산된 \(E_{{{\text{form}}}}\)는 원자당 − 1.87 eV입니다. 음수 값은 형성이 발열임을 의미하고 MnBr2 단층은 에너지적으로 유리합니다. 또한 MnBr2에 대해 계산된 포논 스펙트럼(그림 1d) 단층은 전체 Brillouin 영역에서 음의 주파수를 나타내지 않아 동적으로 안정적임을 나타냅니다. 또한 계산된 탄성 상수(추가 파일 1:표 S2)는 \(C_{11}> 0\), \(C_{11} C_{22} - C_{12)의 Born-Huang 기준 [53]을 준수합니다. }^{2}> 0\) 및 \(C_{66}> 0\), MnBr2 단층은 기계적으로 안정적입니다. 계산된 면내 강성은 26.98J/m
2
입니다. , MnPSe3의 약 75% (36 J/m
2
) [49], MoS2의 15% 단층(180J/m
2
) [54]. 또한 MnBr2 단층은 MoS2에 비해 더 높은 유연성과 더 큰 인장 변형을 견딜 수 있는 능력을 보여줍니다. 단층(11%) [54]. 이것은 MnBr2의 이온 결합에 기인할 수 있습니다. MoS2의 공유 결합에 대한 단층 단층. 탄성 상수와 관련된 변형 분석은 무게를 견딜 수 있음을 나타냅니다(SI의 세부 정보 참조).
MnBr2의 전자 밴드 구조 단층은 그림 1e에 나와 있으며 MnBr2 단층은 직접 밴드 갭이 3.35eV인 반도체입니다. 가전자대 최대값(VBM)과 전도대 최소값(CBM)은 모두 \(\Gamma\) 지점에 있습니다. 전자 구조에 대한 통찰력을 얻기 위해 Mn-d 및 Br-p 궤도에 대한 예상 밀도(DOS)가 그림 1f에 나와 있습니다. Mn 이온의 5개 d 오비탈은 \(a(d_{{z^{2} }} )\), \(e_{1} (d_{xz} + d_{yz} )\) 및 \( e_{2} (d_{xy} + d_{{x^{2} - y^{2} }} )\) \(C_{{{3}v}}\) 대칭에 따라 그룹화됩니다. 더 나쁜 전하 분석은 각 Mn 원자가 두 개의 인접한 Br 원자에 두 개의 전자를 제공함을 시사합니다. 따라서 하나의 스핀 채널에 있는 5개의 d-오비탈은 Mn
2+
의 5개의 d-전자가 완전히 차지합니다. 이온. 이에 따라 두 개의 Mn
2+
단위 셀의 이온은 d
5
에 있습니다. 자기 모멘트가 5μB인 높은 스핀 상태 /− 5μB , Br
1−
이온은 4p
6
의 낮은 스핀 상태에 있습니다. 무시할 수 있는 자기 모멘트 - 0.02μB (추가 파일 1:그림 S1(a)). Goodenough-Kanamori-Anderson(GKA) 규칙에 따르면 이러한 구성은 항상 반강자성 결합을 제공합니다[55].
Mn의 안정성, 전자 및 자기 특성3 Br8 단층
d
5
를 깨기 위해 Mn 공석이 도입되었습니다. Mn
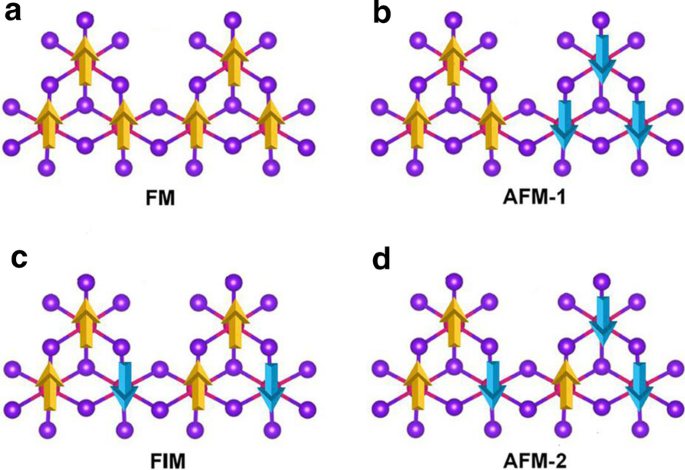
2+
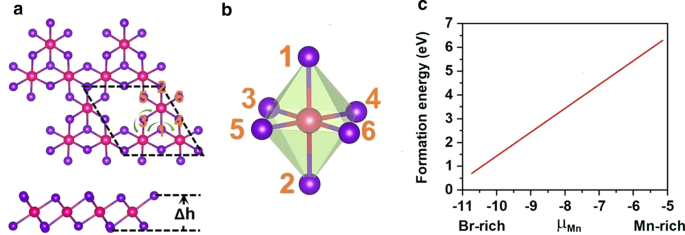
구성 이온. MnBr2의 \(2 \times 2 \times 1\) 슈퍼셀에 단일 Mn 공석이 도입되었습니다. Mn3을 제공하는 단층 Br8 단층. 그림 3a에서 볼 수 있듯이 각 Mn 원자는 4개의 가장 가까운 인접 Mn 원자를 갖고 6개의 Br 원자와 결합하여 왜곡된 팔면체 [MnBr6를 형성합니다. ] 단위. 그림 4에 나타난 5가지 자기상태(NM, FM, FIM, AFM-1, AFM-2)를 고려하였다. 우리의 결과는 FM 상태가 바닥 상태임을 나타내며, 공식 단위당 각각 9.84eV, 32.90meV, 129.85meV 및 97.65meV만큼 다른 4개보다 더 안정적입니다. 최적화된 격자 상수는 여전히 3.95Å입니다. MnBr2과 다릅니다. 단층, Mn3 Br8 단층에는 2가지 유형의 Mn-Br 결합이 있습니다(그림 3b). Mn 원자와 두 개의 중심 Br 원자 사이의 결합(\(d_{{\text{Mn-Br1,2}}}\))은 2.76 Å인 반면, 다른 Mn-Br 결합(\(d_{{\text {Mn-Br3,4,5,6}}}\))는 2.59Å입니다. 두 할로겐화물 평면 사이의 수직 거리는 3.33Å입니다.
<그림>
아 Mn3의 평면도 및 측면도 Br8 단층, \(\Delta h\)는 두 할로겐화물 평면 사이의 수직 거리를 나타냅니다. 원시 세포는 검은색 점선으로 순환됩니다. 녹색 화살표 선은 슈퍼 교환 상호 작용의 두 가지 다른 경로를 보여줍니다. ㄴ 왜곡된 MnBr6의 구조 팔면체. ㄷ Mn의 화학적 포텐셜(μMn)의 함수로서 단일 Mn 공석에 대한 형성 에너지
<그림>
a의 개략도 강자성, b 반강자성-1, c 페리자성 및 d Mn3에 대한 반강자성-2 구성 Br8 단층
Mn 결손 유도의 타당성을 검증하기 위해 우리는 먼저 다음 방정식을 통해 Mn이 풍부한 환경과 Br이 풍부한 환경에서 결손 형성 에너지를 계산했습니다.
여기서 \(E_{{{\text{Mn}}_{{3}} {\text{Br}}_{{8}} }}\) 및 \(E_{{{\text{MnBr}}_ {{2}} }}\)는 Mn3의 총 에너지를 나타냅니다. Br8 및 MnBr2 단층, \(\mu_{{\text{Mn-max}}}\)는 Mn이 풍부한 환경에서 Mn의 화학 포텐셜이며, 이는 벌크 구조에서 Mn 원자의 에너지로 계산됩니다. \(\mu_{ {\text{Mn-min}}}\)는 Br이 풍부한 환경에서 Mn의 화학적 잠재력이며 다음과 같이 계산됩니다.
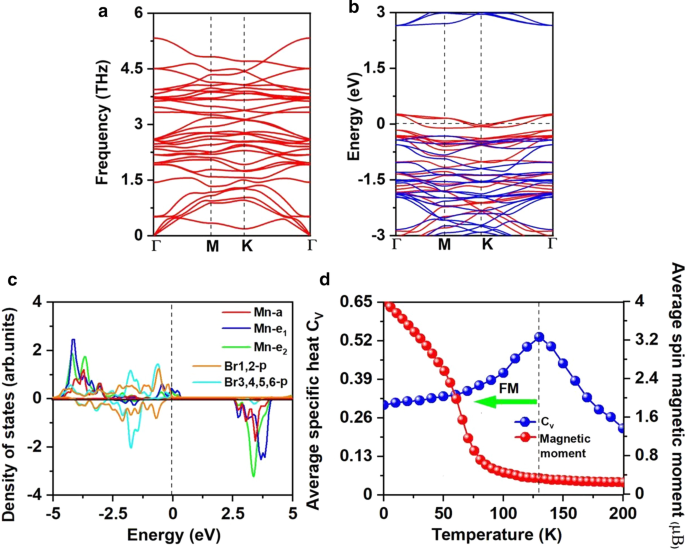
여기서 \(\mu_{{\text{Br-max}}}\)는 Br의 화학 포텐셜이며 기체 상태의 Br 원자 에너지로 계산됩니다. 도 3c에 도시된 바와 같이, Mn-rich/Br-rich 환경에서의 형성 에너지는 Mn vacancy 당 6.30/0.71 eV이며, 이는 Mn vacancy의 형성이 Br-rich 환경에서 에너지적으로 더 유리함을 나타낸다. 실제로 S 공석은 MoS2에서 실험적으로 달성되었습니다. 단층[56]이고 S가 풍부한 환경에서 S 공석의 예측된 형성 에너지는 2.35 eV[57]입니다. 또한, β-FeOOH/PNGN(다공성 질소 도핑 그래핀 네트워크)과 같은 다공성 나노 아키텍처를 구조화하면 상당한 Fe-vacancy를 유발할 수 있으며 [58], Bridgman 방법을 채택하여 Fe vacancy의 정렬을 유도했습니다. 우리는 또한 이러한 방법이 Mn 공석을 유도하는 데 적용되기를 바랍니다[59]. 또한 Mn3의 포논 스펙트럼에는 음의 주파수가 없습니다. Br8 그림 5a에 표시된 단층은 동적 안정성을 입증합니다. 이러한 결과는 Mn 공석을 도입하여 강자성을 가져오려는 우리의 설계를 승인합니다.
<그림>
아 포논 스펙트럼, b 스핀 분해 전자 밴드 구조 및 c Mn3에 대한 Mn-d 궤도 및 Br-p 궤도의 예상 밀도(PDOS) Br8 단층. d Mn 원자의 현장 자기 모멘트와 비열 Cv Mn3에 대한 Heisenberg 모델 기반 온도 함수 Br8 단층. 밴드 구조 및 PDOS에 대한 페르미 레벨은 0eV로 설정
Mn3의 강자성 Br8 FM 슈퍼 교환 상호 작용에 대한 단층 속성. Goodenough-Kanamori-Anderson(GKA) 규칙[55]에 따르면 Mn-Br-Mn 각도가 약 90°일 때 Mn 이온 간의 초교환 상호작용은 FM입니다. 이러한 구성(추가 파일 1:그림 S2)에서 Mn-d 오비탈은 서로 다른 직교 Br-p 오비탈을 사용하여 AFM 커플링하는 경향이 있으므로 간접 Mn-Mn 자기 커플링은 FM이 될 것으로 예상됩니다. 그러나 각 Mn 이온에 MnBr2과 같은 짝을 이루지 않은 전자가 5개 있는 경우 MnBr2에 빈 스핀업 Mn-d 오비탈이 남아 있지 않기 때문에 Mn-Br-Mn 각도가 90°에 가깝지만 단일층, 슈퍼 교환은 AFM입니다. 단층 및 스핀업 d-전자는 인접한 Mn 사이트 사이를 이동할 수 없습니다[60]. Mn3에는 두 개의 서로 다른 슈퍼 교환 상호 작용 경로가 있습니다. Br8 (그림 3a), 둘 다 FM입니다. 하나는 Mn-Br 결합 길이가 2.76Å이고 Mn-Br-Mn 각도가 87.5°인 중심 Br1,2 원자를 포함합니다. 다른 하나는 Mn-Br 결합 길이가 2.59Å이고 Mn-Br-Mn 각도가 95°인 Br3,4,5,6 원자를 포함합니다. Br3,4,5,6 원자의 p 오비탈과 Mn-d 오비탈 사이의 혼성화된 상호작용은 그림 5c에서와 같이 Br1,2 원자를 포함하는 p-d 혼성화의 상호작용보다 강하며, 특히 - 2 eV에서 - 1.4 eV까지입니다. 1.4에서 − 0.9eV인 동안 p -d Br1,2 원자를 포함하는 혼성화가 지배적입니다.
더 나쁜 전하 분석은 각 Mn 원자가 인접한 Br 원자에 8/3의 전자를 제공한다고 제안합니다. 따라서 Mn 이온은 Mn
8/3+
상태. 그림 5c에서 볼 수 있듯이 각 Mn 이온의 13/3 전자는 모두 d-오비탈의 스핀업 채널을 채우고 Br
1-
이온은 4p
6
의 낮은 스핀 상태에 있습니다. . 따라서 각 Mn
8/3+
의 자기 모멘트는 이온은 13/3μB입니다.; Br
1−
의 자기 모멘트 이온은 무시할 수 있습니다(추가 파일 1:그림 S1(b)). d
0
에 대해서도 공극에 의한 강자성 유도를 관찰할 수 있습니다. ZnS 및 ZnO[61, 62]와 같은 시스템에서 단일 공석은 2μB만큼 큰 자기 모멘트를 유발할 수 있습니다. [61]. 각 Mn 이온에 대해 2/3 d-오비탈이 비어 있습니다. \(e_{1}\) 및 \(e_{{2}}\) 궤도의 스핀업 채널은 부분적으로 점유되고 페르미 준위를 가로질러 반금속성을 초래합니다. 반금속 특성은 그림 5b에 표시된 스핀 분해 전자 밴드 구조에서도 관찰할 수 있습니다. 스핀업 채널은 금속성인 반면 스핀다운 채널은 2.97eV의 간접 밴드 갭으로 반도체입니다. VBM/CBM은 \({\text{M}}\)/\(\Gamma\) 지점에서 찾습니다. 밴드 갭 값은 MnP(2.86eV)[63], MnAs(2.92eV)[63], Ni2의 값에 가깝습니다. 아니요2 (2.98 eV) [64], 열적으로 여기된 스핀-플립을 방지하기에 충분히 큽니다. MnBr2와 비교 단층에서 반도체 채널의 VBM과 CBM은 모두 페르미 준위에 더 가까워집니다. CBM은 여전히 Mn 원자가 지배하는 반면 VBM은 새로운 Br1,2 원자가 지배합니다. 한편, 반도체 채널은 직접 채널에서 간접 채널로 전환되고 밴드 갭이 감소합니다. MnCl2에서도 유사한 현상이 관찰되었습니다. H 기능화가 있는 단층 [60].
자화 방향은 자기 이방성 에너지(MAE)에 의해 결정됩니다. 고체의 MAE는 스핀-궤도 커플링(SOC)과 관련된 자기결정 에너지(MCE)와 정자기-정적 쌍극자-쌍극자 상호작용으로 인한 자기 쌍극자 이방성 에너지(MDE)의 두 가지 기여자에서 발생합니다. bcc Fe 및 fcc Ni와 같은 3D 등방성 재료의 MDE는 매우 작습니다. 그러나 자기 모멘트가 큰 전이 금속 원자로 구성된 저차원 재료의 경우 MDE를 무시해서는 안 됩니다[65,66,67]. MCE는 SOC를 고려하여 면내(100 또는 010) 방향과 면외(001) 방향을 따른 자화 에너지의 차이로 정의됩니다. MDE는 면내 자화와 면외 자화 간의 \(E_{d}\) 차이로 얻습니다. 원자 리드베르그 단위의 \(E_{d}\)는 [65, 66]
으로 표시됩니다. $$E_{d} =\sum\limits_{ij} {\frac{{2m_{i} m_{j} }}{{c^{2} }}} M_{ij}$$
여기서 빛의 속도 \(c =274.072\), \(i/j\)는 단위 셀의 원자 위치 벡터이고 \({m}_{i}/{m}_{j}\ ) 원자 자기 모멘트(μB ) 사이트 \(i/j\). 자기 쌍극자 Madelung 상수 \(M_{ij}\)는 다음을 통해 계산됩니다.
여기서 \(R\)은 격자 벡터입니다. 2D 재료에서 모든 \(R\) 및 \(i\)는 면내이므로 두 번째 항은 면외 자화에 대해 0이 되어 양의 \(M_{ij}\ ), 반면 \(M_{ij}\)는 면내 자화에 대해 음수입니다[67]. 따라서 MDE는 전이 금속의 자기 모멘트와 관련이 있으며 항상 평면 내 자화를 선호합니다.
Mn3에 대한 계산된 MCE Br8 단층은 화학식 단위당 − 1.90meV이며(그림 6a), 벌크 Fe(원자당 0.001meV) 및 Ni(원자당 0.003meV)보다 훨씬 크며[68], Rh의 Fe 단층보다 큽니다. (111) (원자당 0.08meV) [69], Mn3 Br8 단층은 열에 안정적입니다. MCE와 방위각 사이의 관계는 다음 방정식으로 설명할 수 있습니다[70].
자기 결정 이방성 에너지(MCE)의 변화 a 방위각 및 b 기준 Mn3 공간에 Br8 단층
여기서 \(A\) 및 \(B\)는 이방성 상수이고 \(\theta\)는 방위각입니다. 피팅 결과는 추가 파일 1에 나와 있습니다. 에스3. 또한 스핀 축이 전체 공간을 회전하는 MCE의 진화가 그림 6b에 나와 있습니다. xy 평면 내의 MCE는 차이가 없으나 xy 평면과 수직인 방향을 따라 최대값에 도달하여 강한 자기 이방성을 확인하였다. MDE는 공식 단위당 − 0.43meV이고 MAE(MCE + MDE)는 공식 단위당 − 2.33meV입니다. 음수 값은 용이한 자화 축이 면내 방향을 따른다는 것을 나타냅니다. MDE는 자화 방향을 변경하지 않지만 향상시킵니다. 또한 Mn3의 MAE Br8 단층은 MnBr2보다 훨씬 큽니다. 단층, 우리 디자인의 효과를 다시 증명합니다.
FM Mn3에 대해 \(T_{c}\)를 추가로 계산했습니다. Br8 Heisenberg 모델을 기반으로 Monte Carlo(MC) 시뮬레이션을 수행하여 단층을 생성합니다. 이는 2D 재료에 대한 \(T_{c}\)를 예측하는 효과적인 방법으로 입증되었습니다 [11, 15, 48, 58, 71,72 ,73,74,75,76]. CrI의 추정 \(T_{c}\)3 단층은 42K(추가 파일 1:그림 S4)[76]로, 실험 측정값[2] 및 이전 계산 결과[15, 58, 71, 72, 74, 76]와 잘 일치하여 의 정확도를 증명합니다. 우리가 채택한 방법. 가장 가까운 이웃(NN) 자기 상호작용을 포함하는 스핀-해밀턴은 다음과 같이 설명됩니다.
$$H =- \sum\limits_{i,j} {JM_{i} M_{j} }$$
여기서 \(J\)는 NN 자기 교환 매개변수이고, \(M_{i/j}\)는 Mn 이온의 자기 모멘트이며 Monte Carlo 방법을 기반으로 하는 스핀 편극 전자의 수에 가까운 적분 [71, 77 , 78], \(i\) 및 \(j\)는 NN 쌍의 Mn 이온을 나타냅니다. 자기 결합 매개변수 \(J\)는 FM과 AFM 상태 간의 에너지 차이를 통해 다음과 같이 계산됩니다.
NN Mn 이온의 계산된 \(J\)는 1.01meV입니다. 양수 값은 FM 커플링을 선호함을 나타냅니다.
NN Mn 이온의 계산된 \(J\) 및 20,000 자기 모멘트 벡터를 포함하는 \(100 \times 100 \times 1\) 슈퍼셀을 채택하여 MC 시뮬레이션을 수행했습니다. 각 온도에서의 시뮬레이션은 10
5
동안 지속됩니다. 단계. 각 자기 모멘트 벡터는 모든 방향으로 무작위로 회전합니다. 그림 5d는 \(C_{{_{V} }} ={{\left( {\left\langle {E^{2} } \right\rangle - \left\langle E \ right\rangle^{2} } \right)} \mathord{\left/ {\vphantom {{\left( {\left\langle {E^{2} } \right\rangle - \left\langle E \right \rangle^{2} } \right)} {K_{B} T^{2} }}} \right. \kern-\nulldelimiterspace} {K_{B} T^{2} }}\) 온도, 여기서 Mn3에 대해 130K의 \(T_{c}\)를 얻었습니다. Br8 액체 질소 온도(77 K)보다 높은 \(C_{v}\)의 피크 위치와 CrI3의 \(T_{c}\)의 피크 위치를 찾아 단층 (45 K) [2] 및 Cr2 Ge2 테6 (28 K) [3], CrX3 (X =F, Cl, Br) (36 ~ 51 K) [11], CrXTe3 (X =Si, Ge) (35.7 K, 57,2 K) [48]. 우리의 계산은 FM Mn3 Br8 단층은 액체-질소 온도보다 높은 MAE 및 퀴리 온도를 갖는다.
Mn3 Br8 이축 변형 및 캐리어 도핑 하에서 단층
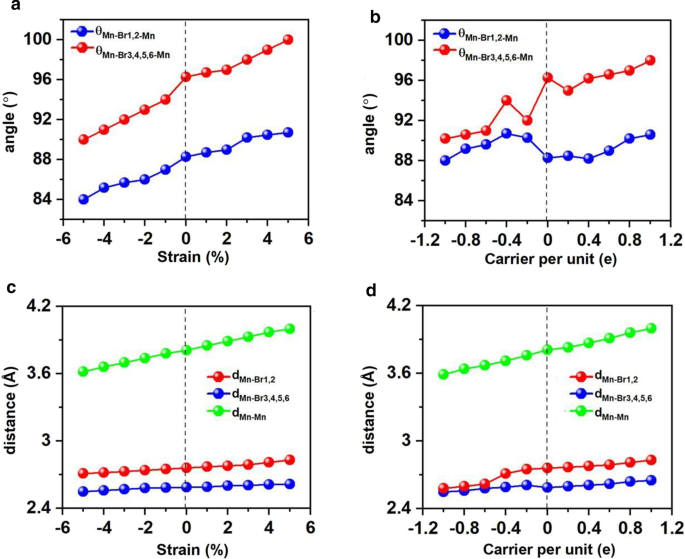
변형 공학은 많은 2D 재료에 적용할 수 있으며 결합 길이 및 각도와 같은 구조적 매개변수를 변경하고 전자 및 자기 특성을 조정하는 데 효과적인 것으로 입증되었습니다. 이러한 맥락에서 우리는 Mn3을 조사했습니다. Br8 - 5% ~ 5% 범위의 이축 변형 하에서 단층. Mn3 Br8 - 5 ~ 5%의 이축 변형 하에서 단층은 FM을 유지하고 원자 자기 모멘트는 거의 변하지 않습니다. 그림과 같이 도 7a 및 c, 2개의 Mn 원자와 Br1,2 원자 사이의 각도(θMn-Br1,2-Mn )는 84°–90°이며, 변형률이 증가함에 따라 증가하고 점차적으로 90°에 접근합니다. Br3,4,5,6 원자를 포함하는 Mn–Br–Mn 각도(θMn-Br3,4,5,6-Mn ) 90°에서 100° 범위에서 점차적으로 90°에서 벗어납니다. 따라서 서로 다른 직교 Br-p 궤도를 통해 매개되는 Mn 이온 간의 슈퍼 교환 상호 작용은 여전히 FM입니다.
<그림>
적용된 이축 변형 및 캐리어 도핑과 관련하여 두 Mn과 Br 원자 사이의 각도 변화, Mn과 Br 원자 사이의 거리, 가장 가까운 이웃 Mn 원자 사이의 거리. a의 변형 각도 및 c 이축 변형에 대한 거리, b의 변형 각도 및 d 캐리어 도핑에 대한 거리. 캐리어 도핑의 양수 및 음수 값은 각각 전자 및 정공 도핑을 나타냅니다.
Mn-Mn 및 Mn-Br 거리는 변형률이 -5%에서 5%로 변경됨에 따라 단조롭게 증가합니다. 이에 따라 그림 8a에 표시된 이축 변형률 하에서 교환 매개변수는 이축 변형률이 -5%에서 5%로 변경됨에 따라 감소하고 -5% 이축 변형률에서 가장 큰 값(1.18meV)에 도달합니다. Mn3의 퀴리 온도 Br8 –5% 이축 변형률 아래의 단층은 160K입니다(그림 9a). Particularly, the Mn-Br bonds under the increasing tensile strain become longer, and the angles of Mn-Br3,4,5,6-Mn deviate from 90°, which are the main reason why the FM super-exchange interaction becomes weaker. Consequently, the Curie temperature decreases. It is similar with CrPTe3 and FePS3 monolayers [79]. Additionally, the MDE decreases with the increasing strain (Additional file 1:Fig. S5(b)); the MAE under –1% biaxial strain is the largest (–3.04 meV). The –5–5% strain does not cause large structural deformation for Mn3 Br8 monolayer, and the morphology of its band structures hardly changes. Mn3 Br8 monolayer keeps to be half-metallic. Both VBM and CBM in the semiconducting spin-channel move upward slightly to the higher energy as shown in Figs. 8c and 10; the band gap increases slowly with the increasing biaxial strain to 3.12 eV under 5% biaxial strain.
Variations of a the exchange parameter and b magnetic anisotropy energy (MAE) for Mn3 Br8 monolayer with respect to the applied biaxial strain and carrier doping. The variations of valence band maximum (VBM), conduction band minimum (CBM), and band gap in the semiconducting channel for Mn3 Br8 monolayer with respect to c the applied biaxial strain and d carrier doping ranging. Positive and negative values of the carrier doping represent the electron and hole doping, respectively
On-site magnetic moments of Mn atoms and the specific heat Cv as function of temperature based on Heisenberg model for Mn3 Br8 monolayer a under -5% biaxial strain, with b 0.2e, c -0.6e, and d -0.8e carrier doping per formula unit. Positive and negative values represent the electron and hole doping, respectively
아 –j Spin-resolved band structure for Mn3 Br8 monolayer under biaxial strain from -5% to 5%. The green arrow denotes the indirect band gap
Electron/hole doping always leads to VBM/CBM moving away from the Fermi level. Our calculations show that Mn3 Br8 monolayer with –1–1e (~ \(1.7 \times 10^{14} {\text{cm}}^{{ - 2}}\)) carrier doping per formula unit is still FM; the atomic magnetic moment of each Mn ion is still 13/3μB. As shown in Fig. 7b and d, with carrier doping from –1e to 1e per formula unit, the Mn-Br-Mn angles involving Br3,4,5,6 atoms are about 90° ~ 98°; the Mn-Br1,2-Mn angles are about 88° ~ 90°. The Mn–Mn and Mn-Br1,2 distances increase with the increasing electron doping. Mn3 Br8 monolayer with 0.2e and 0.4e carrier doping has larger magnetic exchange parameter (Fig. 8a). The Curie temperature at 0.2e electron doping is largest of 140 K (Fig. 9b). Additionally, with –1e ~ 0.2e doping, the MAE is along in-plane directions; the MDE decreases with the increasing electron doping. Under 0.4e doping, the MCE turns to be positive with the value of 0.41 meV per formula unit; the MAE is only 0.01 meV per formula unit with taking the MDE into account (Additional file 1:Figs. S5(a) and (b)). With 0.6e, 0.8e and 1e doping, the PMA (perpendicular magnetic anisotropy energy) is 1.70, 2.42, and 5.13 meV, respectively, large enough for spintronic applications (Fig. 8b).
Additionally, Mn3 Br8 monolayer with carrier doping of –1e ~ 1e per formula unit maintains to be half-metallic. Its band gap in the semiconducting spin-channel increases/decreases slightly with the increasing electron/hole doping as shown in Fig. 8d; the positions of the VBM and CBM do not change. Exceptional, Mn3 Br8 monolayer turns to be FM spin-gapless semiconductors (SGS) with the metallic spin-channel opening up a very small energy gap (0.07 eV) under –0.6e and –0.8e hole doping; its Fermi level locates in the band gap region (Fig. 11b and c, more clearly figures are presented in Additional file 1:Figs. S6(a) and (b)). Correspondingly, electrons may be easily excited from the valence band to the conduction band with a small input of energy, which simultaneously produces 100% spin polarized electron and hole carriers. The Curie temperature at –0.6e and –0.8e hole doping is 110 K (Fig. 9c and d), higher than liquid-nitrogen temperature (77 K). Considering with that the charge density modulation of \(10^{13} \sim10^{15} {\text{cm}}^{ - 2}\) was already achieved experimentally [80,81,82], our predicted properties of Mn3 Br8 monolayer with carrier doping are also experimentally approachable.
아 –j Spin-resolved band structure for Mn3 Br8 monolayer with carrier doping from -1e to 1e per formula unit. Positive and negative values represent the electron and hole doping, respectively. The green arrow denotes the indirect band gap
결론
In summary, the stability, electronic, and magnetic properties of Mn3 Br8 monolayer have been carefully investigated. Our results show that Mn3 Br8 monolayer is FM half-metal with 130 K Curie temperature and with 2.97 eV band gap for the semiconducting spin-channel. Plus, the magnetic moment of each Mn ion is 13/3μB; the MAE is –2.33 meV per formula unit. The Mn3 Br8 monolayer is designed by inducing single Mn vacancy in the \({2} \times {2} \times {1}\) supercell of MnBr2 monolayer to break the AFM coupling d
5
구성. The feasibility of forming the Mn vacancy and the dynamical, mechanical stability of Mn3 Br8 monolayer have been comprehensively confirmed. Additionally, Mn3 Br8 monolayer under biaxial strain –5% ~ 5% is still FM half-metal with 2.71 ~ 3.12 eV band gap for the semiconducting spin-channel, whose Curie temperature under –5% biaxial strain is 160 K. Both biaxial strain and carrier doping make the MAE increase, which turns to be perpendicular to the plane under electron doping. With 0.8e and 0.6e hole doping, Mn3 Br8 monolayer turns to be spin-gapless semiconductor (SGS) with band gap of 0.07 eV. Our calculations demonstrate Mn3 Br8 monolayer as FM half-metal with high Curie temperature, and having large MAE and large magnetic moment, and tunable electronic and magnetic properties under the applied biaxial strain and carrier doping.
데이터 및 자료의 가용성
현재 연구 중 생성 및/또는 분석된 데이터 세트는 합당한 요청이 있는 경우 교신 저자에게 제공됩니다.