첫 번째 원리 계산에서 새로운 오각형 Si/C 복잡성은 리튬 이온 배터리의 유망한 양극 재료로 잠재적으로 응용할 수 있을 것으로 예상됩니다. penta-siligraphene(P-Si2 C4 )은 C 원자만으로 구성된 펜타 그래핀보다 우수합니다. 전자 밴드 구조 분석은 빈 C-2pz P-Si2의 상태 C4 Li의 전자를 수용하고 안정화할 수 있는 공간을 제공하여 Li 저장을 에너지적으로 유리하게 만듭니다. 결과적으로 4개의 Li 원자는 P-Si2의 하나의 화학식 단위에 저장될 수 있습니다. C4 , 1028.7 mAhg
−1
의 이론적 중량 Li 저장 용량에 해당 . Li가 흡착된 P-Lix의 금속 전자 구조 시2 C4 뿐만 아니라 매우 작은 Li 이동 에너지 장벽은 배터리의 빠른 충전/방전 성능에 유리합니다. P-Si2에서 Li 흡착 상호작용에 대한 메커니즘 C4 논의된다. 이러한 결과는 고성능 리튬 이온 배터리를 위한 2차원 Si/C 복합 양극 재료를 설계하기 위한 새로운 전략을 보여줍니다.
<섹션 데이터-제목="배경">
배경
현재 상용화된 리튬 이온 배터리(LIB)의 상대적으로 낮은 에너지 밀도는 상용 전기 자동차(EV)의 요구 사항을 충족하기 어렵고 전기 자동차 산업 발전에 큰 과제가 되고 있습니다[1, 2]. LIB의 에너지 밀도를 높이려면 전극 재료의 용량을 개선해야 합니다. 매우 우수한 사이클링 성능으로 인해 흑연은 가장 널리 사용되는 양극 재료이지만 이론적인 중량 용량(372 mAhg
−1
) 상대적으로 낮다[3, 4]. 반면에 실리콘은 약 4200 mAhg
−1
의 매우 높은 이론 중량 용량을 가지고 있습니다. [5], 그러나 사이클링 성능은 완전히 리튬화된 상태에서 최대 420%까지 매우 큰 부피 팽창으로 인해 열악합니다[6]. 실리콘 양극과 탄소 양극을 모두 활용하려면 Si/C 복합 양극을 설계하는 것이 학문적으로나 기술적으로 중요합니다.
C 원자가 부분적으로 Si 원자로 대체된 2차원(2D) 그래핀과 같은 층상 물질인 실리그라핀은 첫 번째 원리 계산[7,8,9,10]에서 안정적인 2D 물질로 처음 예측되었으며 실험 [11, 12]에서 성공적으로 준비되었습니다. Linet al. 2D SiC 시트는 용액 박리 기술을 통해 준비될 수 있음을 보여주었습니다[11]. 그들은 또한 quasi-2D SiC2를 성공적으로 준비했습니다. 몇 달 동안 공기 중에서 보존될 수 있는 시트[12]. 나중에 첫 번째 원리 계산에 따르면 실리그라핀은 1520 mAhg
−1
의 이론적 용량을 제공하는 유망한 양극 물질입니다. 및 1286 mAhg
−1
g-SiC5용 및 g-SiC2 , 각각 [13]. siligraphene 양극은 흑연 양극의 높은 사이클 안정성과 실리콘 양극의 고용량을 물려받는 것으로 나타났습니다. 예상되는 높은 Li 저장 용량은 sp에서 Si 원자의 변화와 관련된 siligraphene 단층과의 향상된 Li 흡착 상호 작용에 기인합니다.
2sp
3
-같은 [13]. 그러나 전자 구성은 sp에서 변경됩니다.
2sp
3
-like는 siligraphene에 Li 흡착 동안 명백한 구조적 변화를 동반합니다. 이는 LIB의 양극 재료로 사용되는 실리그라핀의 사이클링 성능에 좋지 않습니다. 더 나은 솔루션은 이미 sp가 있는 Si/C 복합 재료를 설계하는 것입니다.
3
-같은 전자 구성.
탄소에는 sp로 구성된 여러 유형의 동소체가 있습니다. , sp
2
, 및 sp
3
혼성화 또는 이들의 조합. 매우 안정적인 sp
2
흑연 탄소의 큰 π-결합 전자 구성은 그래핀에서 약한 Li 흡착 상호 작용을 담당합니다. 단층 그래핀에 Li가 흡착되면 Li에서 그래핀 층으로 전하 이동이 일어난다[14]. 그러면 Li는 양전하를 띠고 매력적인 쿨롱 상호작용에 의해 그래핀 층에 결합된다. 그러나 그래핀 층의 Li로부터의 과잉 전하는 그래핀의 큰 π-결합을 깨뜨리고 이는 에너지적으로 바람직하지 않다. 그 결과, 단일층 그래핀에 대한 Li 흡착은 신체 중심면의 응집 에너지보다 음으로 높은 흡착 에너지(bcc ) 리튬 이온 배터리에서 허용되지 않는 상 리튬 금속. 결과적으로 깨끗한 그래핀 단층을 사용한 리튬 저장은 허용되지 않습니다[15]. 대안적으로, 경질 탄소 재료는 흑연 탄소 재료에 비해 훨씬 더 높은 Li/Na 저장 중량 용량을 제공합니다[16,17,18]. 경질 탄소 재료는 sp
2
및 sp
3
탄소 원자 [19]. 경질 탄소 재료의 더 높은 Li 저장 중량 측정 용량이 sp
3
전자 구성?
sp로 알려진 펜타 그래핀
2
-sp
3
하이브리드 2D 탄소 동소체[20]는 첫 번째 원리 계산[21]에서 Li/Na 이온 배터리의 유망한 양극 재료로 예측되었습니다. 펜타 그래핀은 2차원 탄소 동소체로서 기존의 벌집 구조의 그래핀에 비해 리튬 흡착 거동이 훨씬 강하다. 이 다른 Li 흡착 거동도 sp와 관련이 있습니까?
3
-펜타 그래핀의 전자 구성과 유사합니까? 대답이 '예'인 경우 그 이면의 내재적 메커니즘은 무엇입니까?
펜타-그래핀은 역학적으로 안정적인 탄소 동소체로 예측되지만, 그 응집 에너지는 전 세계적으로 가장 안정적인 상(흑연 또는 그래핀)에 비해 훨씬 높습니다. 펜타 그래핀의 응집 에너지는 단일층 육방정계 그래핀보다 원자당 약 0.9 eV 높기 때문에[20, 22], 펜타 그래핀을 산업적으로 대규모로 제조하기가 매우 어렵습니다(가능한 경우). 그러나 애노드 재료로 응용하려면 대규모 제작이 매우 중요합니다. 좌굴은 실리신에서 발견되므로 Si는 sp에서 더 안정적입니다.
3
- sp보다 혼성화 유사
2
[23,24,25] 반면 C 원자는 sp를 선호합니다.
2
2D 구조의 혼성화; sp
3
펜타-그래핀의 구조에서 Si 원자를 가진 C 원자는 에너지적으로 선호될 것입니다. 우리는 이 구조를 펜타실리그라핀이라고 부릅니다. 최근의 실험은 오각형 Si 기반 나노 리본이 Ag(110) [26]에서 성장될 수 있음을 보여주었으며, 이는 Si 기반 오각형 구조의 형성이 실험적으로 가능함을 보여줍니다.
이론적으로 펜타실리그라핀(P-SiC2 ) Lopez-Bezanilla et al. 그리고 그들은 P-SiC2 p-p-σ 및 p-p-π 전자 밴드의 수직 순서의 부분적인 반전을 나타냅니다[27]. 나중에 P-SiC2의 전자 수송 특성 penta-graphene 및 penta-CN2과 연구 및 비교 [28]. 흥미롭게도 P-SiC2의 전자 수송 성능이 변형 공학을 통해 조정할 수 있으며 단축 압축 변형이 단층 penta-SiC2의 정공 이동성을 향상시킬 수 있다고 예측되었습니다. 최대 1.14 × 10
6
cm
2
V
−1
s
−1
[29]. 구조의 유사성에도 불구하고 penta-siligraphene은 penta-graphene과 비교하여 다른 수송 특성을 가지고 있습니다. Hu et al.에 의해 발견되었습니다. penta-graphene의 열전도율은 stretch에 의한 표준 monotonic reduction을 나타내는 반면 penta-SiC2 특이한 non-monotonic 위아래 동작을 가지고 있습니다 [30]. 펜타실리그라핀의 이러한 흥미로운 특성은 구조에 있는 Si 원자의 전자 및 화학적 특성과 밀접하게 관련되어 있습니다. 또한 Si 원소 자체가 Li 흡착을 향상시키는 데 도움이 되는 것으로 밝혀졌는데, 이는 실리콘에 대한 Li 흡착 상호작용이 그래핀에 대한 상호작용에 비해 훨씬 강하기 때문입니다[31, 32]. 따라서 penta-siligraphene이 LIB의 양극 재료로 사용될 수 있는지 아는 것이 흥미로울 것입니다.
이 연구에서 우리는 첫 번째 원리 계산을 사용하여 펜타-실리그래핀의 리튬 이온 저장 거동을 조사하고 펜타-실리그래핀에 의해 리튬 이온이 어떻게 저장될 수 있는지에 대한 메커니즘에 대해 구체적으로 논의합니다. 우리는 penta-siligraphene의 열역학적 안정성에서 연구를 시작한 다음 Li 흡착의 본질적인 상호 작용에 대한 자세한 분석을 시작합니다. 마지막으로 LIB의 양극재로서의 펜타실리그라핀의 성능에 대해 논의합니다.
계산 방법
이 작업의 모든 계산은 밀도 함수 이론(DFT)에 기반한 Vienna Ab initio Simulation Package(VASP)[33]를 사용하여 수행됩니다. Perdew-Burke-Ernzerhof(PBE)에 의해 매개변수화된 일반 기울기 근사(GGA) 교환 및 상관 함수와 결합된 PAW(Projector Augmented Wave) 방법[34, 35]이 사용됩니다[36]. 평면파의 차단 에너지는 모든 계산에서 450 eV로 선택됩니다. 격자 매개변수와 이온 위치는 완전히 이완되고 최종 힘은 0.02 eV/Å로 수렴됩니다. 전자 밴드 구조는 Heyd-Scuseria-Erznerhof(HSE06) hybrid functions[37]으로 계산됩니다. 상태 밀도(DOS) 계산은 0.05 eV의 스미어링 너비로 가우스 스미어링 방법으로 스미어링됩니다. Monkhorst-Pack [38] k -포인트 샘플링이 사용되며 k의 밀도 -메쉬가 0.05 Å
−1
보다 두껍습니다. 초기 분자 역학(AIMD) 시뮬레이션 및 0.03 Å
−1
의 경우 다른 계산을 위해. 원자 전하 분포는 Bader 전하 분석으로 분석됩니다[39]. 리튬 이온 이동 경로는 CINEB(Climbing Image nudged Elastic band) 방법으로 최적화되었습니다[40]. 흡착 에너지 E광고 계산:
여기서 E호스트 , E리 , 및 E호스트 + 리 호스트 펜타-실리그라핀 재료, Li 원자 및 Li 흡착 호스트 각각의 총 에너지, n 는 펜타실리그라핀에 흡착된 Li 이온의 수를 나타낸다. 흡착 에너지에 대한 반 데르 발스(vdW) 상호 작용의 영향은 Becke-Jonson 감쇠가 있는 DFT-D3 방법을 사용하여 테스트됩니다[41]. 흡착 에너지 외에도 평균 Li 삽입 전위(vs Li
+
/Li)는 V에서 Li 금속(bcc상)의 흡착 에너지와 응집 에너지의 차이로부터 직접 얻을 수 있습니다. 이브 =− (E광고 − ELi − 응집력 ), 에너지와 전위의 단위로 eV와 V를 각각 선택하면
결과 및 토론
펜타실리그라핀의 구조 및 안정성
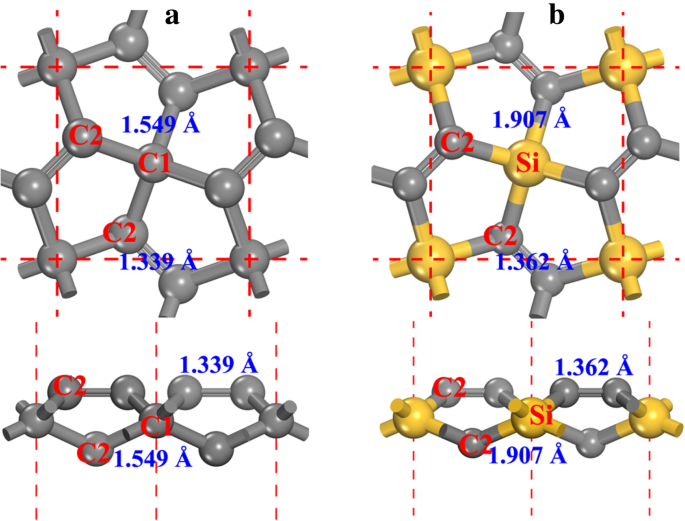
펜타 그래핀의 구조(그림 1a 참조, P-C6로 표시됨) 이 백서 다음에서) P-421나 대칭(공간군 번호 113). 최적화된 격자 상수는 a =b =3.636 Å, 이전 결과와 일치[20, 21]. 구조에서 두 가지 유형의 탄소 원자, 즉 4-배위 탄소(그림 1a에서 C1로 표시)와 3-배위 탄소(그림 1a에서 C2로 표시)를 찾을 수 있습니다. 탄소 원자의 국부적 기하학에서 우리는 C1이 sp임을 알 수 있습니다.
3
-C2가 sp인 동안 하이브리드화됨
2
- 하이브리드 처럼. C2 원자는 sp로 간주되지만
2
-like hybridized[20], 이중 C2-C2 결합 특징은 C2 원자의 화학적 특성을 그래핀의 화학적 특성과 다르게 만듭니다. 이에 대해서는 이 백서의 다음에서 자세히 논의될 것입니다. P-C6에서 C1 원자를 Si 원자로 교체 구조, penta-siligraphene이 형성되고(그림 1b 참조, 최적화된 구조의 결정 정보 파일은 추가 파일 1:보충 재료의 SI-1에 제공됨) P-Si2 C4 이 문서의 다음에서. Si 원자의 원자 반경이 C 원자보다 크므로 P-Si2의 격자 상수 C4 (아 =b =4.405 Å)는 P-C6보다 큽니다. , 다른 보고된 결과와 잘 일치하지만 [27,28,29,30].
<사진>
아 펜타그래핀과 b의 볼과 스틱 모델 펜타실리그라핀. 평면도(위쪽)와 측면도(아래쪽)가 모두 표시됩니다. 회색 및 노란색 구체는 각각 C 및 Si 원자입니다. 4- 및 3-배위 탄소 원자는 각각 C1 및 C2로 표시됩니다. 본드 길이도 각 본드와 함께 표시됩니다.
상대적 열역학적 안정성을 평가하기 위해 표 1은 C, Si 및 C/Si 착물의 다른 동소체의 응집 에너지를 나타냅니다. 비록 펜타-그래핀(P-C6 )는 초기 분자 역학(AIMD) 시뮬레이션[20], P-C6의 응집 에너지로부터 1000 K에서 안정적인 것으로 나타났습니다. (− 8.24 eV·atom
−1
)은 단층 그래핀(− 9.14 eV·atom
−1
)에 비해 훨씬 높습니다. ). 이것은 P-C6의 대량 생산을 보여줍니다. 매우 어려울 것입니다. 한편, P-Si2의 응집에너지는 C4 (− 7.26 eV·atom
−1
)는 가장 안정적인 동소체인 g-Si2에 비해 0.2 eV만 더 높습니다. C4 (− 7.46 eV·atom
−1
), penta-siligraphene의 준비가 P-C6에 비해 훨씬 쉬울 수 있음을 보여줍니다. . P-Si2의 구조적 안정성을 확인하기 위해 C4 , P-Si2의 포논 분산 곡선 C4 Γ-point(0.0039 THz 또는 0.13 cm
−1
) 근처의 작은 영역에서 작은 가상 주파수가 발견되지만 ), 이러한 작은 가상 주파수(1 cm
−1
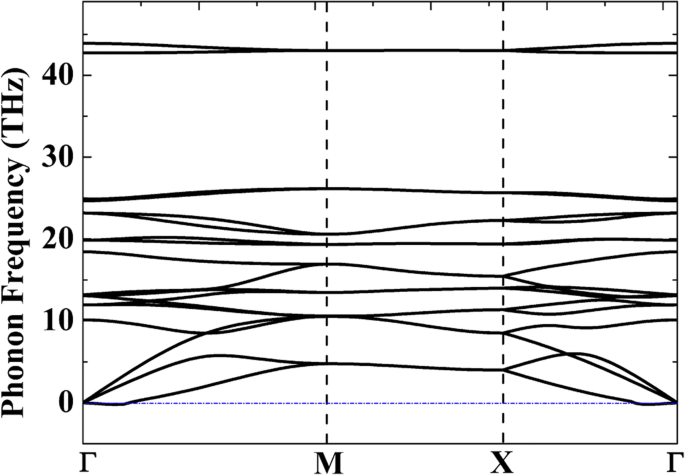
이하)가 일반적으로 받아들여지기 때문에 시스템이 동적으로 안정적이라고 믿을 수 있습니다. ) 시뮬레이션의 인공물일 수 있습니다[42]. 가상 주파수는 게르마넨[43] 및 아르세넨[44]과 같은 다른 동적으로 안정적인 2D 재료에서도 보고되었습니다. 계산의 정확도를 높이거나 다른 계산 방법을 사용하는 것과 같은 기술 처리를 적용하면 이러한 가상 주파수를 제거할 수 있습니다.
<그림>
2D P-Si2의 포논 분산 곡선 C4 선형 응답 이론에서 계산된 단층
또한 AIMD 시뮬레이션을 수행하여 P-Si2의 구조적 안정성을 평가합니다. C4 고온에서. AIMD는 1000 K, 1500 K, 2000 K 및 2500 K의 온도에서 표준 앙상블에서 3 × 3 및 4 × 4 슈퍼셀을 사용하여 수행됩니다(추가 파일 1:그림 S1 참조). 추가 파일 1:그림 S2 및 S3은 P-Si2의 원자 구성을 나타냅니다. C4 각각 3 × 3 및 4 × 4 슈퍼셀을 사용하여 서로 다른 온도에서 AIMD 시뮬레이션이 끝날 때. 보인 바와 같이 20ps 시뮬레이션 시간 동안 2000 K의 높은 온도에서는 오각형 원자고리가 변하지 않고 구조가 2000 K의 높은 온도를 견딜 수 있음을 보여주지만, 반면에 심각한 구조적 변형 스냅샷에 육각형 고리(추가 파일 1:그림 S2d 참조) 및 기타 결함(추가 파일 1:그림 S3d)이 나타나 구조가 2500 K에서 파괴되었음을 나타냅니다. 2 C4 2500 K에서 g-Si2 C4 (육각형 고리[13]로 구성됨)는 5상 P-Si2보다 더 안정적입니다. C4 , 표 1에 주어진 응집 에너지와 일치합니다. 이러한 결과는 P-Si2의 구조적 안정성이 C4 P-C6에 비해 훨씬 안정적입니다. , 1000 K의 온도만 견딜 수 있습니다.
펜타실리그라핀의 리튬 흡착
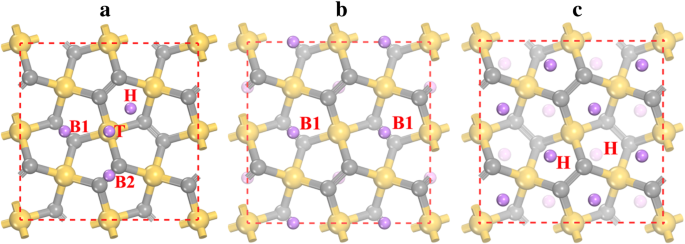
penta-siligraphene P-Si2에 대한 Li 흡착을 연구하기 위해 C4 , 다른 Li 흡착 사이트가 고려되며 이완 후 4개의 안정적인 흡착 사이트(그림 3a 참조)가 발견될 수 있습니다. 안정적인 Li 흡착 사이트는 Si 원자의 상단 사이트(T로 표시), Si2의 중공 사이트(H로 표시)입니다. C3 오각형 고리, 그리고 하층(B1)과 상층(B2)에 있는 두 개의 C2 원자 사이의 브리지 사이트. 이 사이트에서 Li 이온 흡착의 선호도는 표 2에 제시된 흡착 에너지로 특징지을 수 있습니다. 결과는 가장 안정적인 Li 흡착 사이트가 - 1.922 eV의 흡착 에너지를 갖는 B1 사이트임을 보여줍니다. 반면에 H 사이트의 흡착 에너지(- 1.905 eV)는 B1 사이트에 매우 가깝습니다. Li 흡착 에너지는 흡착 높이로도 표시되는데, 흡착 에너지가 낮을수록 흡착 높이가 작기 때문입니다(표 2 참조). Li 흡착 과정이 시작될 때 Li 이온은 가장 안정적인 B1 위치에 머무르는 것이 좋습니다. 모든 B1 사이트가 점유된 후(Li2의 화학량론에 해당) 시2 C4 및 그림 3b 참조), Li 이온은 H 사이트에 머물기 시작합니다. B1과 H site 사이의 거리가 매우 작기 때문에(~ 1.5 Å) B1과 H site에서 Li 이온과 강한 반발 상호작용이 일어난다. 결과적으로 B1 사이트의 Li 이온은 인근 H 사이트로 반발되어 B1 사이트가 비어있는 반면 모든 H 사이트는 Li4 상태로 점유됩니다. 시2 C4 (그림 3c 참조). Li 흡착 에너지에 대한 vdW 상호 작용의 영향도 테스트되었으며 결과는 표 2의 괄호 안에 나와 있습니다. 표시된 바와 같이 vdW 상호 작용은 - 0.12에서 - 0.17 eV까지 다양한 흡착 위치에 대한 흡착 에너지에 기여합니다. vdW 상호 작용이 Li 흡착에 유리함을 보여줍니다.
<그림>
a의 리튬 흡착 사이트 penta-siligraphene 표면과 b의 가장 안정적인 구조의 원자 배열 리2 시2 C4 및 c 리4 시2 C4 . 노란색(가장 큰), 회색(중간 크기) 및 보라색(가장 작은) 구체는 각각 Si, C 및 Li 원자입니다. H, T, B1 및 B2는 Li 흡착 사이트를 나타냅니다.
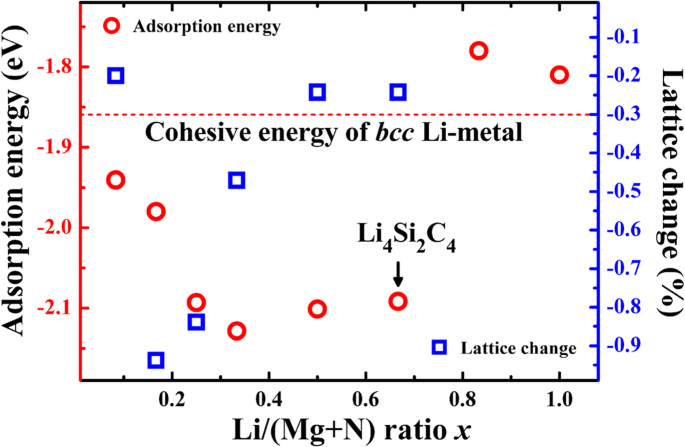
Li 흡착 에너지와 bcc의 응집 에너지 비교 상 Li 금속(− 1.86 eV·atom
−1
), Li 흡착이 전기화학적으로 활성인지 아닌지를 판단할 수 있다. 흡착 에너지가 Li 금속의 응집 에너지보다 낮으면 Li 이온 흡착이 유리하고 흡착은 양의 방전 전위에 해당합니다. 표 1에서 볼 수 있듯이 B1과 H 사이트의 흡착 에너지는 - 1.86 eV보다 낮으며, 이는 B1과 H 사이트가 모두 Li 저장을 위한 전기화학적 활성 사이트임을 보여줍니다. Li 저장 용량을 평가하기 위해 다양한 리튬 이온 농도에서 Li 흡착 에너지 x (Li/(Si + C) 비율)을 계산하여 bcc의 응집 에너지와 비교합니다. 리 메탈. 그림 4에서 보는 바와 같이 Li/C 비율이 x일 때 Li 흡착 에너지는 - 1.86 eV보다 낮습니다. P-Si2의 하나의 단위 셀에 흡착된 4개의 Li 원자에 해당하는 2/3보다 작습니다. C4 및 1028.7 mAhg
−1
의 이론적 중량 측정 용량 (리4 시2 C4 ). 용량 곱하기 출력 전압과 같은 배터리의 에너지 밀도는 리튬 저장 용량에 비해 더 중요합니다. 좋은 양극 물질은 흡착 에너지로부터 얻을 수 있는 상대적으로 낮은 전기화학적 포텐셜을 가져야 합니다. 평균 전위는 약 0.1–0.2 V이며, 이는 상대적으로 낮고 전체 배터리 시스템의 더 높은 출력 전압에 유리합니다. 또한 그림 4는 다양한 Li 흡착 농도에서 격자 상수 변화를 보여줍니다. 도시된 바와 같이, P-Si2의 격자상수 C4 Li 흡착시 약간 수축합니다. 농도가 x일 때 1/6일 때 격자 변화가 - 0.94%로 가장 큰 값에 도달하여 충방전 과정에서 체적 변화가 매우 작음을 나타냅니다. 작은 부피 변화는 P-Si2의 구조를 유지하는 데 도움이 됩니다. C4 사이클링 중에 안정적입니다.
<그림>
P-Si2에서 계산된 Li 흡착 에너지 C4 표면(빨간색 주기, 왼쪽 축) 및 Li 흡착 농도(Li/(C+Si) 비율)의 함수로서의 격자 상수 변화(파란색 사각형, 오른쪽 축). bcc의 응집력 비교를 위해 상 Li 금속도 포함됩니다.
Li 흡착 시 Penta-siligraphene의 전자 구조 분석
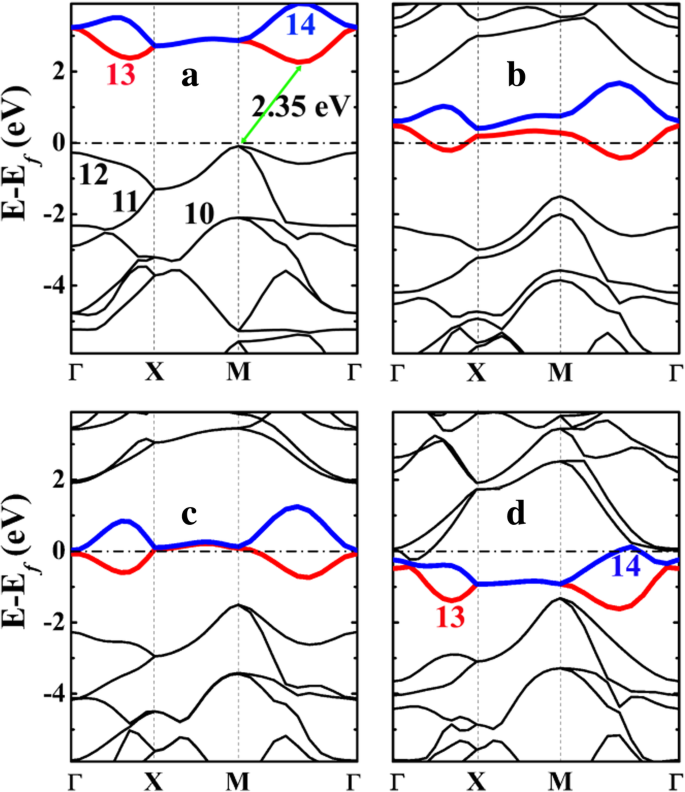
펜타실리그라핀(P-Si2 C4 ) 및 리튬화 상태가 그림 5에 나와 있습니다. P-Si2 C4 간접 밴드 갭이 약 2.35 eV인 반도체로 펜타 그래핀 P-C6의 3.46 eV에 비해 훨씬 작습니다. (추가 파일 1:그림 S4 참조). P-Si2의 더 작은 밴드 갭 C4 PC6보다 이는 특히 높은 대칭성 M 및 Γ 지점에서 가장 높은 점유 밴드(그림 5a의 11번 및 12번)의 향상된 분산에서 비롯됩니다. 12번(가장 높은 점유 대역)과 13번(가장 낮은 비점유 대역)의 에너지 대역 사이에 밴드 갭이 열린다. 12번 밴드의 에너지 준위는 M 지점에서 실질적으로 상승하여 페르미 준위를 높이고 차례로 밴드 갭을 감소시킵니다.
<그림>
a의 전자 밴드 구조 펜타실리그라핀 P-Si2 C4 및 리튬화 상태 b P-LiSi2 C4 , ㄷ 피리2 시2 C4 , 및 d 피리4 시2 C4 HSE06에서 계산됨. 페르미 준위는 0 eV로 선택됩니다. a의 10에서 14까지의 숫자 그리고 d 는 밴드 번호를 나타내며 밴드 번호 13과 14는 각각 빨간색과 파란색으로 강조 표시됩니다. 밴드 라벨링은 VASP 코드를 따르며, 밴드 라벨링은 가전자대 및 전도대를 참조하고 코어 전자는 라벨링에 포함되지 않습니다.
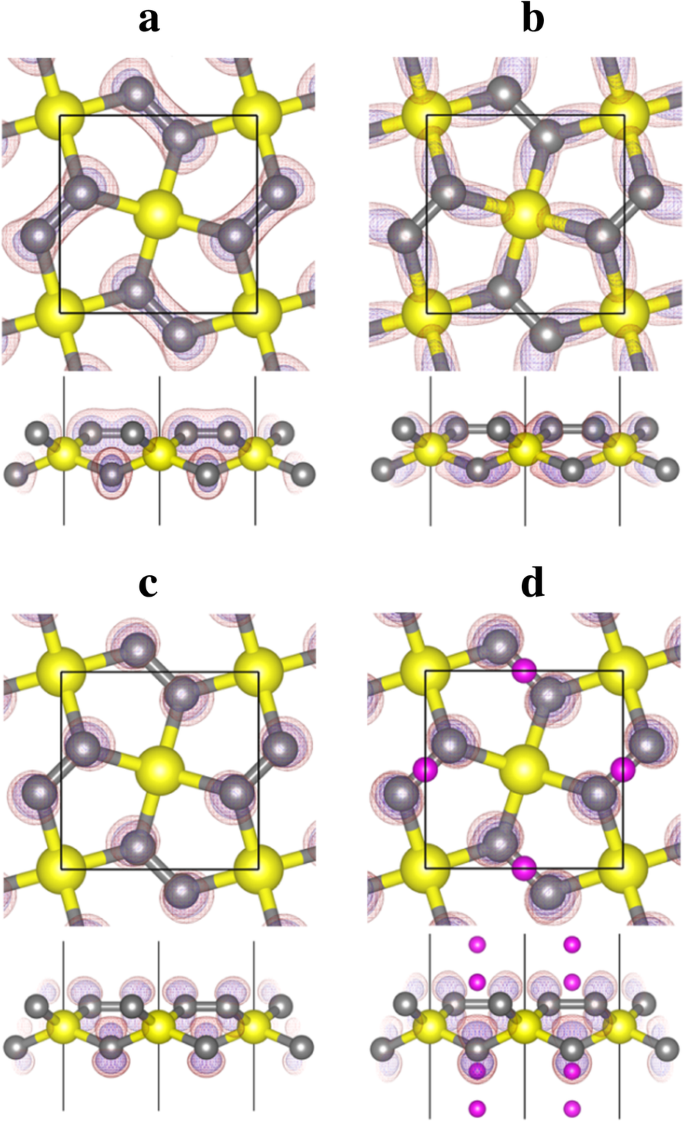
그림 6에 표시된 밴드 10-14에 투영된 전하 밀도(파동 함수)의 모양 분석에서 밴드 번호 12가 C와 Si 사이에 형성된 σ-결합의 결합 상태에 해당함을 알 수 있습니다. 원자, 밴드 번호 13(및 14)은 2-p에 해당합니다. z C 원자의 상태. 빈 C-pz 상태는 Li 흡착으로부터 전자를 수용하고 안정화할 수 있는 공간을 제공하여 Li 흡착 과정을 에너지적으로 유리하게 만듭니다.
<그림>
밴드 a 에 대한 밴드 분해 전하 밀도 등고선 10번, b 12번 및 c 펜타실리그라핀(P-Si2)의 13번 C4 , 그림 5a) 및 밴드 d 리튬화 펜타-실리그라핀(Li4 시2 C4 , 그림 5d). 노란색(큰), 회색(중간 크기) 및 보라색(작은) 구체는 각각 Si, C 및 Li 원자입니다. 전하 밀도 등고선은 투명한 빨간색으로 표시됩니다(등가곡면 값 0.02 e/Å
3
). ) 및 파란색(등가곡면 값 0.01 e/Å
3
) ) 색상
P-Si2에서 가장 높은 점유 에너지 밴드의 향상된 분산 C4 이는 두 가지 요인에 기인할 수 있습니다. 첫째, 탄소 단독 펜타-그래핀 PC6에서와 비교하여 12번 밴드(C-Si σ-결합)를 차지하는 전자와 양으로 하전된 Si 원자 사이의 쿨롱 인력 . Bader 전하 분석은 Si 원자가 penta-siligraphene P-Si2에서 양전하를 띠고 있음을 보여줍니다. C4 . 표 3에서 볼 수 있듯이, siligraphene 또는 penta-siligraphene에서 Si 원자의 Bader 전하는 약 1.65 e입니다. , Si 원자는 + 2.35 e로 양전하를 띠고 있음을 보여줍니다. . 한편, 펜타그래핀의 C1 원자(P-C6 )도 양전하를 띠지만 순 전하는 + 0.08 e입니다. . 따라서 공유 결합 상호 작용 외에도 P-Si2에서 C와 Si 원자 사이의 강력한 쿨롱 상호 작용이 발생합니다. C4 , P-C6에 비해 . 이는 페르미 준위 부근의 점유 에너지 대역(12번)의 분산에 유리하다. 둘째, P-Si2의 향상된 좌굴 C4 더 큰 좌굴이 더 많은 sp를 의미하기 때문에 밴드 번호 12(C-Si σ-결합의 결합 상태)의 분산에 기여할 수도 있습니다.
3
- 하이브리드화되고 더 강한 σ-결합이 C-Si 원자 사이에 형성됩니다. 이는 P-Si2의 안정적인 구조적 안정성을 위한 중요한 이유이기도 합니다. C4 P-C6와 비교 . 더 중요한 것은 Si와 C 원자 사이의 좌굴이 Li 흡착과 함께 증가하고 리튬화 상태 P-Li4에서 0.876 Å이 된다는 것입니다. 시2 C4 . 이 상태에서 SiC4의 C–Si–C(102.50° 및 124.59°) 결합각 사면체는 표준 사면체의 108.47°에 가까워지며 sp
3
- Si 원자의 혼성화와 C-Si 결합의 강도는 Li 흡착 시 더 강해집니다. 결과적으로 그림 4에서 볼 수 있듯이 12번 밴드의 분산도 증가된 Li 흡착 농도와 함께 향상됩니다.
penta-siligraphene(P-Si2) 표면에 Li 흡착시 C4 ), Li 원자에서 전하 이동(실제 배터리 작동에서 Li
+
이온은 내부 회로에서 오는 반면 동일한 양의 전자는 외부 회로에서 나옵니다) P-Si2의 탄소 원자 C4 . 결과적으로 초과 전자는 비어 있는 밴드(밴드 번호 13 및 14) 아래로 이동하여 그림 6b–d와 같이 시스템의 금속 전자 구조를 생성합니다. 금속 전자 구조는 P-Si2의 우수한 전자 전도성을 보장합니다. C4 P-Si2를 사용하는 배터리 시스템의 속도 성능에 유익한 충방전 과정 중 양극 C4 양극.
위에서 논의되고 그림 6의 대역 투영 전하 밀도로 표시되는 바와 같이 대역 번호 13과 14는 pz P-Si2에서 C 원자의 상태 C4 . 이러한 빈 밴드는 Li 흡착에 매우 중요합니다. 양으로 하전된 Li 이온과 음으로 하전된 P-Si2 사이의 쿨롱 인력 외에도 C4 기질, C-p를 차지하는 전자 z 상태는 양전하를 띤 Si 원자(13번과 14번 밴드의 에너지 준위를 아래로 이동하여 기질의 총 에너지를 낮춤)에 대해 강한 쿨롱 인력을 갖습니다. 결과적으로 P-Si2에 Li 흡착 C4 정력적으로 더 유리합니다. P-Si2의 단위 셀로 C4 4개의 C 원자를 포함하고 4개의 Li 원자가 P-Si2에 흡착될 수 있을 것으로 예상됩니다. C4 표면. C-p 뒤에 z 상태가 완전히 점유됨, P-Si2에 더 많은 Li 원자 흡착 C4 에너지적으로 불리할 것입니다. 이것은 그림 4에 제시된 계산된 흡착 에너지와 일치합니다.
P-Si의 리튬 이온 마이그레이션 역학2 C4
P-Si2의 속도 성능 C4 양극은 전자 전도 및 리튬 이온 확산 역학에 의해 결정됩니다. 위에서 논의한 바와 같이 순수한 P-Si2의 전자 구조는 C4 절연체이므로 Li 농도가 낮은 경우에도 Li 흡착에 따라 자발적으로 금속이 됩니다. 따라서 음극재로 응용하기에는 전자전도도가 충분히 좋아야 한다. 그러면 penta-siligraphene에 대한 Li-ion 확산이 속도 조절 단계가 됩니다. P-Si2의 구조로 C6 펜타그래핀(P-C6 ) Li 이온이 매우 빠르게 확산되는 [21] P-Si2에서 Li 이온 확산이 예상됩니다. C6 매우 빠를 수도 있습니다.
배터리의 속도 성능은 충전 상태(SOC)와 밀접한 관련이 있습니다. 즉, 리튬 이온 확산은 리튬 흡착 농도에 따라 달라집니다. 다른 SOC에서 Li 이온 확산 역학을 평가하기 위해 두 가지 극한 농도, 즉 희석 Li 이온과 희석 Li 공석이 고려됩니다. 묽은 리튬 이온 확산 시뮬레이션을 위해 하나의 Li 이온이 P-Si2 슈퍼셀에 흡착됩니다. C4 . "Penta-siligraphene에 대한 Li 흡착" 섹션의 위 논의에서 Li 이온은 그림 7과 같이 Li 농도가 낮을 때 B1 사이트에 머무르는 것을 선호합니다. 구조의 대칭성을 고려할 때 하나의 Li 이동 경로( 그림 7에서 Path-1로 표시됨)을 찾을 수 있으며 Path-1은 P-Si2에서 완전한 2D Li 확산 네트워크를 형성합니다. C4 표면. P-Si2의 묽은 리튬 이온 이동 에너지 장벽 C4 along the NEB method optimized pathway is about 0.117 eV, which is smaller than that of on P-C6 (0.17 eV, Path-II) [21] and g-Si2 C4 (0.548 eV) [13]. When the SOC becomes 50%, namely, the material is discharged into the state of Li2 시2 C4 , all Li ions still occupy the B1 sites (see Fig. 3b) and thus the Li-ion diffusion pathway is the same as the case of dilute Li ion. These results show that Li-ion diffusion can be very fast at the beginning half of the discharge process.
The dilute Li-ion migration pathway and the corresponding energy profile along the pathway on P-Si2 C4 . The gray (middle sized), golden, and purple spheres are C, Si, and Li atoms, respectively. The red arrows illustrate the two-dimensional diffusion networks
For the case of dilute Li vacancy, we remove one Li ion from the fully lithiated state Li4 시2 C4 and create one dilute Li vacancy in the supercell. As discussed above, Li ion prefers to occupy the H site when the Li concentration is high (see Fig. 3c). Therefore, three different Li vacancy migration pathways are considered, as shown in Fig. 8a. Path-1 refers to the Li vacancy migration from the H site to its neighboring H site across the top of a Si atom (across T site). Path-2 corresponds to the pathway across the top of the middle point of the C2–C2 dimer at the top layer (across B2 site). Path-3 is the pathway along the C2–C2 dimer at the down-layer (across B1 site). The energy profiles along the optimized pathways are given in Fig. 8b. As is seen, the Li-ion migration energy barriers along these pathways are very low, particularly for Path-3 (0.052 eV). The energy profiles along Path-1 and Path-2 are slightly asymmetric, because of the large relaxation of the Li ions when one Li vacancy is created. The extremely low energy barrier along Path-3 is reasonable, since Path-3 crosses B1 site (energetically most favorable adsorption site). However, Path-3 alone is not able to form a complete Li diffusion network on the surface of the P-Si2 C4 . Therefore, Path-1 or Path-2 must take part in the diffusion process, and the overall energy barrier for dilute Li vacancy migration is 0.155 eV or 0.165 eV. Although higher than dilute Li-ion migration (0.117 eV), the energy barrier for dilute Li vacancy migration is also very small compared with that for P-C6 (0.25 eV, Path-II’) [21] and g-Si2 C4 (0.233 eV) [13]. As the Li migration energy barriers in P-Si2 C4 are always lower than those in P-C6 and g-Si2 C4 (both dilute Li ion and dilute Li vacancy), it is expected that the rate performance of the P-Si2 C4 is the best one among the three similar anode candidates.
아 Dilute Li vacancy migration pathways and b the corresponding energy profiles on fully lithiated P-Li4 시2 C4 . The gray (middle sized), golden, and purple spheres are C, Si, and Li atoms, respectively. The large green sphere represents the dilute Li vacancy. The thick/thin arrows indicate fast/slow migration pathways which form the two-dimensional diffusion networks
결론
In summary, based on first principles calculations, we predicted that 2D pentagonal Si/C compound P-Si2 C4 can be potentially used as anode materials for LIBs. Phonon dispersion data confirmed the dynamic stability of the P-Si2 C4 structure at ground state, while AIMD simulation shows that the structure of the P-Si2 C4 can be stable at temperatures as high as 2000 K. The unique 2D buckled pentagonal structure promotes special empty C-2pz states that facilitate Li adsorption on the surface of the P-Si2 C4 , which offers a gravimetric Li storage capacity of 1028.7 mAhg
−1
. The calculated dilute Li-ion/Li vacancy migration energy barriers show that Li-ion diffusion on the surface of the P-Si2 C4 can be faster than both the pentagonal graphene (P-C6 ) and the honeycomb-structured siligraphene. The metallic electronic structure of the lithiated P-Lix 시2 C4 ensures good electronic conductivity of the material as electrodes. These advantages are crucial features to the P-Si2 C4 as a promising anode material for LIBs. In summary, our first principles study offers a novel strategy to design high-performance Si/C complexity for the application in LIBs.