hGC33-변형 및 Sorafenib-Loaded 나노입자는 Wnt 신호 경로를 억제하여 상승적인 항간종 효과를 나타냅니다.
초록
종양 특이적 억제제의 전달은 암 치료의 도전입니다. 항체로 변형된 나노입자는 로딩된 약물을 특정 종양 관련 항원을 과발현하는 종양 세포에 전달할 수 있습니다. 여기에서 우리는 간세포 암종에서 과발현되는 막 단백질인 글리피칸-3(GPC3 +)에 대한 항체 hGC33으로 변형된 소라페닙이 로딩된 폴리에틸렌 글리콜-b-PLGA 고분자 나노입자를 구성했습니다. 우리는 hGC33 변형 NP(hGC33-SFB-NP)가 GPC3
+
를 표적으로 한다는 것을 발견했습니다. 간세포암종(HCC) 세포는 HCC 세포 표면의 GPC3에 특이적으로 결합하여 Wnt 유도 신호 전달을 억제하고 G0/1에서 cyclin D1 발현을 하향 조절하여 HCC 세포를 억제하여 상피를 억제하여 HCC 세포 이동을 약화시킵니다. 간엽 전환. hGC33-SFB-NP는 각각 GPC3 분자와 함께 Ras/Raf/MAPK 경로 및 Wnt 경로를 억제함으로써 HCC 세포의 이동, 주기 진행 및 증식을 억제하였다. hGC33-SFB-NP는 생체 내에서 간암의 성장을 억제하고 종양이 있는 마우스의 생존율을 개선했습니다. 우리는 hGC33이 HCC 세포에 대한 SFB-NP의 표적화를 증가시킨다는 결론을 내렸습니다. hGC33-SFB-NP는 Wnt 경로와 Ras/Raf/MAPK 경로를 차단하여 HCC의 진행을 상승적으로 억제합니다.
소개
글리피칸3(GPC3)은 글리세로포스파티딜이노시톨 앵커를 포함하는 메커니즘을 통해 세포 표면에서 발현되는 헤파란 설페이트 프로테오글리칸입니다[1]. GPC3는 발달 동안 다양한 조직에서 발현되지만, 그 발현은 프로모터 영역의 DNA 메틸화에 의해 대부분의 성인 조직에서 적어도 부분적으로 억제된다[2]. 그러나 GPC3 단백질은 간세포암종(HCC) 환자의 약 70%에서 과발현되고[3, 4] Wnt 리간드와의 상호작용을 통해 고전적인 Wnt 신호 전달[5]을 자극하며, 이는 Wnt/frizzled 결합을 촉진하여 HCC 성장을 촉진합니다[6]. . 고전적인 Wnt 신호 전달 경로의 활성화는 HCC의 악성 형질전환과 관련된 가장 빈번한 사건 중 하나입니다[7, 8]. Wnt 신호 전달을 증가시키는 GPC3의 능력을 기반으로, 우리는 GPC3의 과발현이 고전적인 Wnt 경로를 자극하여 HCC 성장을 촉진한다는 가설을 세웠습니다.
GPC3(524-563)의 C-말단 에피토프를 인식하는 GPC3(524-563)(hGC33)의 C-말단 부분의 에피토프를 인식하는 치료용 단일클론항체는 HepG2 및 Huh- 마우스에서 7 [9, 10]. hGC33은 상보적 결정영역 이식(complementary decision-region 이식)을 통해 인간화되었으며, HepG2 이종이식에 대한 항암 효과는 hGC33만큼 효과적이며, hGC33은 임상에서 사용되고 있다[11]. 이러한 결과는 hGC33이 중요한 항종양 활성을 가지며 항-GPC3 치료가 Wnt 및/또는 기타 신호 전달 경로를 차단함으로써 HCC 세포의 증식 및/또는 생존을 직접적으로 억제할 것임을 시사합니다.
그러나 간세포암종의 복잡한 병인으로 인해 GPC3 단독 차단 효과는 제한적이다[12]. 따라서 간세포암종의 발병기전의 상세한 기전을 탐색하고 간세포암종의 진단 및 예후에 대한 유망한 바이오마커를 식별하는 것은 효과적인 치료 표적을 제공하고 환자의 예후를 개선하는 데 도움이 될 수 있습니다. 항-GPC3 항체는 화학요법제에 대한 간세포암종의 감수성을 증가시키기 위해 제안되었다[13]. 표준 화학 요법 약물과 함께 hGC33의 항종양 활성이 최근에 평가되었습니다[14]. 소라페닙은 RAF/MEK/ERK가 매개하는 세포 신호 전달 경로를 차단하여 종양 성장을 억제함으로써 종양 세포의 증식을 직접 억제할 수 있는 새로운 경구 표적 간세포암종 약물입니다[15, 16]. 그러나 생체 내 소라페닙의 비표적 분포와 간세포암종에서 비정상적으로 활성화된 Wnt 신호는 약물의 효과를 제한하고 부작용을 증가시킨다[14,15,16]. Wnt는 Frizzled 계열의 수용체에 결합하여 세포 증식, 세포 사멸 및 세포 이동을 조절하는 세포 내 신호 전달을 활성화하고 HCC와 같은 많은 종양에서 약물 내성을 유발합니다[17,18,19].
HepG2 이종이식 모델에서 hGC33과 소라페닙의 조합은 소라페닙 단독보다 종양 성장 억제에 더 효과적이며[15], 고분자 나노운반체를 이용한 약물 전달은 암 치료에서 많은 관심을 받고 있다. 항암 약물이 탑재된 나노캐리어는 생체 내 비특이적 분포 및 비특이적 분해를 방지하고 약물 생체 이용률 및 항종양 표적화를 개선하며 약동학 및 치료 평가를 단순화할 수 있습니다[15, 16]. 다양한 고분자 기반 나노 입자에서 폴리(락트산 공 글리콜산)(PLGA) 기반 제형은 이상적이고 안전한 약물 운반체로 간주됩니다[17, 18]. 이와 관련하여 폴리(에틸렌 글리콜)-b -폴리(d,l-락티드-코-글리콜리드)(PEG-b -PLGA)는 폴리에틸렌 글리콜과 PLGA 공중합체를 기본으로 하며 가수분해 후에도 안전하고 독성이 없으며 미국 식품의약국(FDA)의 승인을 받았습니다[19,20,21,22]. 따라서 PEG-b 나노캐리어를 정맥 주사 -PLGA 공중합체는 표적 전달을 달성하고 효능을 향상시키는 유망한 전략입니다. 또한, HCC 세포막에 GPC3 분자에 대한 특정 항체 hGC33을 적용하면 시험관 내 및 생체 내에서 표적화하는 나노약물의 전달을 개선할 수 있을 뿐만 아니라 [18, 19], Wnt 및/또는 GPC3와 연결된 기타 신호 경로를 차단하고 억제할 수 있습니다. 암세포의 증식 및/또는 생존, 그리고 상승적인 항종양 활성을 달성할 수 있습니다.
이 연구에서 우리는 hGC33 항체 변형 공중합체 PEG-b -PLGA 나노입자는 in vivo 및 in vitro에서 간세포암종으로의 소라페닙(hGC33-SFB-NP) 전달을 용이하게 할 수 있으며, 약물 약동학을 변화시키는 HCC 활성 표적화를 통해 간세포암 치료의 효율성을 향상시킬 수 있습니다. 입자 크기, 제타 전위, 입자 형태, 약물 포획 효율, 약물 로딩 용량 및 시험관내 약물 방출에 따라 표적 NP를 포괄적으로 특성화하였다. 시험관내 표적화 능력은 HepG2 간암 세포의 세포 흡수를 특징으로 합니다. hGC33-SFB-NP의 간세포암종에 대한 생체분포 및 상승적 치료 효과를 소라페닙과 SFB-NP를 비교하여 평가하였다. 우리의 결과는 hGC33-SFB-NP가 GPC3
+
를 표적으로 할 수 있음을 보여주었습니다. 간세포암종 Wnt 및 Ras/Raf/MAPK 신호 경로를 억제하여 세포 주기 진행, 세포 증식 및 종양 침범을 억제하고 간암의 진행을 상승적으로 억제할 수 있습니다.
자료 및 방법
자료
hGC33 항체, 3-(4,5-디메틸티아졸-2-일)-2,5-디페닐테트라졸륨 브로마이드(MTT), 4',6-디아미디노-2-페닐인돌, 디히드로클로라이드(DAPI), 5,5',6 ,6'-테트라클로로-1,1',3,3'-테트라에틸-벤즈이미다졸릴 카르보시아닌 요오드화물(JC-1) 및 디메틸 설폭사이드(DMSO)는 Sigma-Aldrich, Inc.(St. Louis, MO)로부터 입수했습니다. 1-(3-디메틸아미노프로필)-3-에틸-카르보디이미드 염산염 및 N -hydroxysuccinimide는 Qiyun Biotech(Guangzhou, China)에서 입수했습니다. bicinchoninic acid(BCA) 단백질 정량 키트, coumarin-6 및 Annexin V-FITC/PI 세포자멸사 검출 키트는 Beyotime Biotechnology(중국 상하이)에서 구입했습니다. PEG-b -PLGA-말레이미드 이중블록 공중합체(mal-PEG-b) -PLGA; 25,000–30,000 Da, PLGA, LA:GA, w/w; PEG, 13-15%)는 Polyscitech(West Lafayette, IN, USA)에서 구입했습니다. PI3K 항체 키트(9655#), p-Akt 항체 키트(9916#), mTOR 항체 키트(9964#), Bcl-2 family 항체 키트(9942#), apoptosis 항체 키트(9915#), 2차 염소 항바이러스제 토끼 및 항-마우스 항체는 Cell Signal Technology(Danvers, MA, USA)에서 구입했습니다. 사이클린 B1 및 사이클린 의존성 키나제는 Abcam Biological Technology(미국)에서 구입했습니다. phospho-Rb, cyclin D1, 체크포인트 키나제 1(CHK1), P53, 인산화된 유방암 감수성 유전자 1(p-BRCA1), RAD51, 시토크롬 C 및 기질 메탈로프로테이나제(MMP2 및 MMP9)에 대한 항체는 Abcam(USA)에서 구입했습니다. . 기타 모든 분석 등급의 화학 물질, 시약 및 용매는 표준 공급업체로부터 입수했으며 추가 정제 없이 사용되었습니다.
세포와 동물
HCC 세포주 HepG2(American type culture collection(Manassas, VA, USA)에서 입수)를 10% FBS(HyClone, Logan, UT, USA)가 보충된 DMEM 배지(Invitrogen, Carlsbad, CA, USA)에서 배양하였다. 5% CO의 습한 분위기에서 U/ml 페니실린 및 80μg/ml 스트렙토마이신2 37°C에서 20~22g(5~6주) 무게의 BALB/C 누드 마우스는 Nanjing Junke Biotechnology Co., Ltd.(중국)에서 제공했습니다. BALB/C 누드 마우스는 SPF 방에서 사육되었습니다. 모든 동물 관리 및 처리는 Anhui University of Science and Technology의 동물 관리 요구 사항에 따라 수행되었습니다. 모든 실험 프로토콜은 Anhui University of Science and Technology의 동물 실험 윤리 위원회에서 검토 및 승인되었습니다(승인 번호:2019dw013).
NP 준비
NP를 준비하려면 mal-PEG-b -PLGA 및 SFB 또는 쿠마린-6을 칭량하고 유기상(3:2 v/v 디클로로메탄/아세톤)에 용해시켰다. 용액을 연속적인 볼텍싱과 함께 한 방울씩 폴리비닐 알코올(PVA) 용액(5% w/v)에 첨가하였다. 혼합물을 얼음 위에서 프로브 초음파 분쇄기(550W의 출력, 8회)로 간헐적으로 초음파 처리하여 오일-물 에멀젼을 생성했습니다. 에멀젼을 자기 교반과 함께 PVA(1% w/v) 용액에 첨가하였다. SFB 및 쿠마린-6 NP는 8000rpm에서 30분 동안 원심분리하여 수집하고 Milli-Q 물로 3회 세척했습니다.
항체 hGC33에서 말레이미드와 유리 설프히드릴 잔기의 반응에 의해 형성된 티오에테르 결합에 의해 hGC33-NP를 생성하기 위해, hGC33 항체를 말레이미드 작용화된 NP와 5:1의 몰비로 혼합하였다(hGC33:mal-PEG-b -PLGA) 및 4°C에서 16시간 동안 계속 교반하면서 인큐베이션했습니다. hGC33은 항체 hGC33의 설프히드릴 그룹과 PEG 사슬의 말레이미드 그룹의 반응을 통해 NP에 접합되었습니다. 접합되지 않은 항체는 Sepharose CL-4B 컬럼을 통해 통과시켜 제거했습니다. 효율적인 단백질 접합은 BCA 키트(Thermo Fisher Scientific, Waltham, MA, USA)로 확인되었습니다.
나노입자(NP)의 특성화
형태, 입자 크기, 캡슐화 효율성(EE) 및 NP의 안정성
나노입자의 형태는 투과전자현미경(TEM, H-600; Hitachi, Tokyo, Japan)으로 평가하였다. 약물이 없는 hGC33-NP 및 약물이 없는 NP는 브롬화칼륨을 사용하여 FTIR 분광광도계(Thermo Nicolet, Madison, WI, USA)로 기록했습니다. NP의 평균 입자 크기와 제타 전위는 20°C에서 Malvern Zetasizer ZEN3600 Nano ZS(Malvern Instruments, Malvern, UK)로 특성화되었습니다. 약물 캡슐화 효율(EE) 및 약물 로딩 함량(LC) 효율을 한외여과에 의해 평가하였다. 샘플을 한외여과 장치(100kMWCO; Sartorius, Göettingen, Germany)에 로드하고 4°C에서 25분 동안 8000rpm으로 원심분리하여 유리 약물을 제거했습니다. 각 시료의 동일한 부피를 아세토니트릴에 녹여 약물의 총량을 확인하였다. 농도는 고성능 액체 크로마토그래피로 측정했습니다. 흡수 파장은 266nm였습니다. 다음 공식을 사용하여 NP의 약물 EE(%)를 계산했습니다:(포획된 약물의 중량/약물의 총 중량) × 100%. LC(%)는 (캡슐화된 약물의 중량/NP의 중량) × 100%로 계산되었습니다. 실온에서 나노입자의 안정성을 이해하기 위해 미리 결정된 시점(0.5, 1, 2, 4, 8, 12시간, 16시간, 20시간, 24시간)에서 NP 크기의 변화를 동적 광산란(DLS)으로 평가했습니다. h) 25°C에서
약물의 체외 약물 방출 및 세포 흡수 분석
NPs의 약물은 10kDa의 분자량 컷오프를 가진 투석 백을 사용하여 조사되었습니다. 간단히 말해서, 1mL의 NP를 투석 백(MWCO 8,000–10,000Da; Spectrum Labs Inc., CA, USA)에 넣었습니다. 투석 백을 PBS에 담그고 25°C에서 자기 교반기로 교반했습니다. NP의 약물 방출 프로필은 100mL의 0.2M 인산완충식염수(PBS, pH =7.4)에서 7일 동안 측정되었습니다. 시료의 약물 농도는 고성능 액체 크로마토그래피로 측정했습니다. 후속 연구에서는 hGC33-SFB-NP와 동일한 입자 크기를 갖는 hGC33-coumarin 6-NP를 사용하여 hGC33-SFB-NP의 표적화를 평가하였다. HepG2(GPC3
+
) 및 Li-7(GPC3
−
) hGC33-coumarin 6-NP와 함께 0.5, 2 또는 4시간 동안 37°C, 5% CO2,에서 인큐베이션했습니다. 각기. 공동 배양된 세포를 세척하고 10분 동안 4% 포름알데히드로 고정했습니다. 세포 핵을 5μg/mL Hoechst 33,342로 15분 동안 염색하여 세포 내 NP를 찾습니다. 세포내 나노입자 이미지를 분석하기 위한 공초점 현미경은 공초점 현미경(Olympus FV1000; Olympus Corporation, Tokyo, Japan)으로 분석되었습니다.
체외 세포 효과
유리 hGC33(Ab), 유리 SFB, hGC33-null-NP 또는 hGC33-SFB-NP의 세포독성은 MTT 분석을 사용하여 결정되었습니다. HepG2(GPC3
+
) 세포 및 Li-7(GPC3
−
) 대수 단계의 세포를 웰당 4000개 세포의 밀도로 96웰 플레이트에 시딩한 다음 5% CO2에서 37°C에서 48시간 동안 인큐베이션했습니다. . 세포를 hGC33, 유리 SFB, hGC33-null-NP 또는 hGC33-SFB-NP로 37°C, 5% CO2에서 48시간 동안 처리했습니다. . 일정한 시간 동안 공배양한 후, 항세포 증식 활성은 기술된 바와 같이 MTT 분석에 의해 결정되었다[20]. 각 웰의 흡광도는 490nm에서 측정되었으며 SPSS 17.0으로 최대 억제 농도 값(IC 50)의 절반을 계산했습니다.
세포 침범 능력 측정
대수 성장기의 세포를 5 × 10
4
의 밀도로 6웰 플레이트에 접종했습니다. 세포/웰을 흡입 헤드로 긁고 무혈청 배양 배지로 교체했습니다. 상처 치유는 대조군과 실험군에서 0시간, 24시간, 48시간에 기록되었습니다. 동시에 Transwell chamber에 같은 수의 세포를 접종하고 free hGC33, free SFB, hGC33-null-NP, hGC33-SFB-NP를 처리하였다. 500 마이크로리터의 10% FBS 배지를 하부 챔버에 추가했습니다. 24시간 동안 배양한 후 Transwell 챔버를 꺼내고 세포를 4% 파라포름알데히드로 고정하고 0.1% 크리스탈 바이올렛으로 염색했습니다. 상처 치유의 크기와 이동 세포의 수를 계산하여 이동 능력을 평가했습니다.
세포 주기 측정
6웰 플레이트에서 밤새 배양한 후 세포를 유리 hGC33(Ab), 유리 SFB, hGC33-null-NP 또는 hGC33-SFB-NP로 24시간 동안 처리한 다음 수집하고 에탄올로 고정했습니다. propidium iodide로 염색한 후 유세포분석을 하고 modifit 3.0(Verity Software House, Topsham, ME)으로 세포주기를 분석하였다.
서양 얼룩
신호 경로의 활성화 상태와 표적 분자의 발현을 평가하기 위해 세포를 6-웰 플레이트에서 밤새 배양하고 유리 hGC33, 유리 SFB, hGC33-null-NP 또는 hGC33-SFB-NP를 24시간 동안 적용했습니다. . 각 처리군의 세포를 채취하여 단백질을 추출하여 측정하였다. 단백질 농도는 BCA 단백질 키트(Biosharp, Hefei, China)로 측정 및 보정되었습니다. 샘플의 단백질은 12개의 알킬 설페이트 폴리아크릴아미드 겔 전기영동으로 분리하고 PVDF 멤브레인으로 옮기고 무지방 우유로 밀봉하였다. 첫 번째 항체(1:1000로 희석)를 4C에서 밤새 인큐베이션하고 두 번째 항체(1:2000)를 실온에서 1시간 동안 인큐베이션했습니다. 밴드는 ECL 기질(Thermo Fisher Scientific Waltham, MA, USA)로 가시화되었고 이미지는 겔 이미지 분석 시스템으로 표시되었으며 β-액틴은 대조군으로 사용되었습니다.
체내 항종양 활동
생체 내 HCC 성장에 대한 유리 hGC33, 유리 SFB, hGC33-null-NP, SFB-NP 및 hGC33-SFB-NP의 억제를 측정하였다. Anhui University of Technology의 윤리 위원회의 동물 건강에 관한 규정 및 지침에 따라 모든 실험은 12시간/12시간 조명이 있는 온도 조절실(23 ± 2 °C)의 우리에 있는 BALB/c 마우스에서 수행되었습니다. 다크 사이클. 5 × 10
6
을 포함하는 50μl 현탁액 살아있는 HepG2 세포를 5주령 암컷 BALB/c 마우스(20~22g)의 오른쪽 복부에 피하 주사했습니다. 종양 부피가 약 50mm에 도달한 경우
3
, 마우스를 무작위로 6개 그룹(각 그룹에 10마리)으로 나누었다. 일반 식염수 대조군 NS(200μL PBS 중 200mg/kg null NP), hGC33-null-NP(200μL PBS 중 hGC33-null-NP, hgc33 =100μg/kg/시간에 해당), 유리 hGC33(hGC33 in 200μL PBS, 100μg/kg/시간), 유리 SFB(SFB 용량:8mg/kg/시간), SFB-NP(SFB 용량:8mg/kg/시간) 및 hGC33-SFB-NP(동등한 SFB =8mg/kg/time, hGC33 =100μg/kg/time)을 꼬리 정맥을 통해 2일마다 10회 주입했습니다. 4일마다 마우스의 체중과 종양 크기를 측정하였다. 종양 부피의 계산 공식은 부피 =0.5 × L × 와
2
, 여기서 L과 W는 각각 종양의 길이와 너비를 나타냅니다. 투여 4주 후 동물을 디에틸 에테르로 마취하고 종양의 크기와 무게를 측정하였다. 또한 종양, 심장, 간, 신장, 폐 및 비장을 제거하고 4% 파라포름알데히드 용액으로 고정하고 파라핀에 포매한 후 절개하고 헤마톡실린과 에오신으로 염색하여 디지털 현미경으로 조직학적 변화를 평가했습니다.
통계 분석
데이터는 평균 ± 표준편차(SD)로 표시되며 SPSS 18.0으로 분산 분석에 의해 평가되었습니다. 쌍별 통계 비교는 양측 스튜던트 t 테스트를 사용하여 수행되었습니다. 차이는 P에 대해 통계적으로 유의한 것으로 간주되었습니다. <0.05.
<섹션 데이터-제목="결과">
결과
NP 및 약물 방출의 특성화 시험관
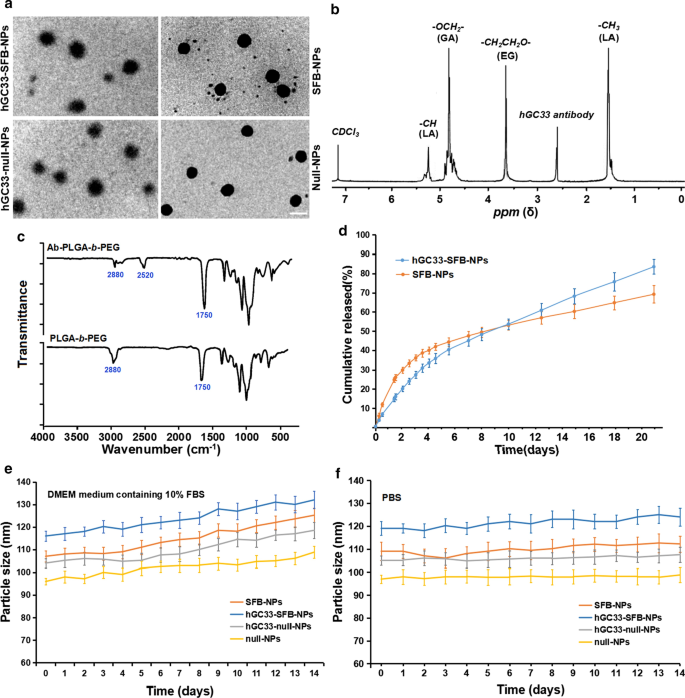
NP의 직경과 표면 특성이 세포 흡수, 약물 방출 및 생체 내 NP 분포에 영향을 미치기 때문에 우리는 해당 매개변수를 사용하여 준비된 NP를 특성화했습니다. SFB-NP, hGC33-null-NP, hGC33-SFB-NP 고분자의 형태, 입도, 입도분포를 그림 1에 요약하였다. 투과전자현미경으로 관찰한 hGC33-SFB-NP의 형태(Fig. . 1a) 가장자리가 흐릿한 단단한 핵을 보여주며, 이는 hGC33이 NP의 표면에 존재함을 나타냅니다. SFB NP, hGC33-null-NP 및 hGC33-SFB-NP의 입자 크기는 100~150nm 범위이며 전형적인 단봉형 입자 크기 분포를 나타냅니다. hGC33-SFB-NP(120.2 ± 10.2nm)의 평균 직경은 hGC33-null-NP, SFB-NP 및 null NP보다 약간 컸습니다(그림 1a, 표 1). hGC33-SFB-NP 및 hGC33-null-NP의 표면에는 단일 표면을 가진 얇은 구형의 불분명한 필름이 존재하여 NP 표면에 항체 hGC33이 존재함을 나타냅니다. hGC33-SFB-NP 및 hGC33-null-NP의 크기가 증가하여 hGC33 필름의 존재를 확인했습니다. hGC33-SFB-NP의 합성은
1
로 확인되었습니다. NP 준비 전 H-NMR(그림 1b). 5.2ppm 및 1.58ppm의 피크는 젖산의 -CH3 양성자에 할당되었습니다. 4.8ppm의 피크는 글리콜산의 -OCH2-에 할당되었습니다. 3.6–3.8ppm의 피크는 PEG 반복 단위의 -CH2CH2O- 양성자에 할당되었습니다. PEG-b의 표면 화학 -PLGA 및 Ab-PEG-b -PLGA NP는 FTIR 분광법으로도 연구되었습니다(그림 2c). PEG-b의 스펙트럼에서 -PLGA 폴리머, 약 1750cm의 강한 밴드
−1
PLGA 사슬에서 카르보닐기(C =O)의 스트레치에서 유래. 2880cm의 밴드
−1
이는 PEG 사슬에서 -CH 그룹의 스트레칭 때문이었습니다. 동시에 2520cm
−1
에서 피크가 나타났습니다. 우리는 hGC33 항체의 -SH 스트레칭 피크에 기인합니다.
1
H NMR 및 FTIR 결과는 항체가 PEG-b의 골격에 이식되었음을 나타냅니다. -PLGA 폴리머.
<그림>
NP의 특성화. 아 NP의 TEM 특성화, 눈금 막대는 100nM을 나타냅니다. ㄴ
1
합성 hGC33-PLGA-b의 H NMR 스펙트럼 -CDCl3의 PEG; ㄷ hGC33-PEG-b의 FTIR 스펙트럼 -PLGA 및 PEG-b -PLGA; d 37°C에서 PBS(pH =7.4)에서 SFB-NP 및 hGC33-SFB-NP의 누적 방출 프로필; 이 14일에 걸쳐 10% FBS를 포함하는 DMEM 배지에서 배양된 NP의 크기 변화; 에 14일 동안 PBS에서 배양된 NP의 크기 변화. SFB, 소라페닙; TEM, 투과 전자 현미경; 나노입자, 나노입자;
1
H NMR,
1
H-핵 자기 공명 분광법; FTIR, 푸리에 변환 적외선 분광기
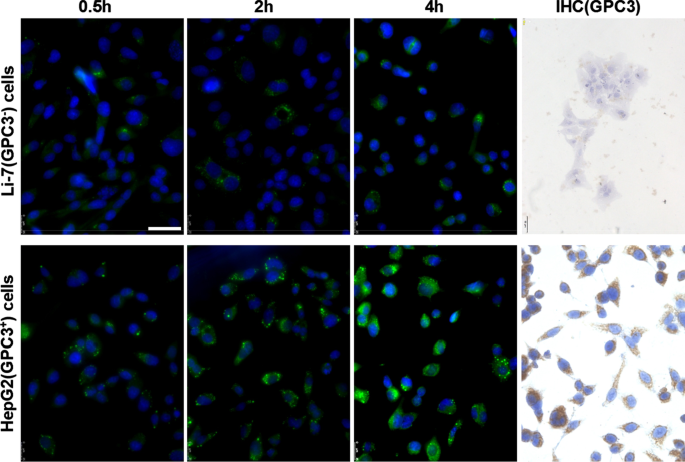
<그림>
Li-7 및 HepG2 세포에서 GPC3의 발현 및 hGC33-coumarin6-NP의 흡수. 배양 플레이트에 접종된 세포를 PBS로 세척하고 DMEM에서 100μg/ml hGC33-coumarin6-NP와 함께 2, 4, 8시간 동안 배양했습니다. 핵을 DAPI로 염색하고, 세포를 고정하고 공초점 레이저 스캐닝 현미경으로 검출하였다. GPC3는 Li-7 세포에서 검출되지 않았지만, 면역세포화학에 의해 검출된 바와 같이 HepG2 세포에서 높게 발현되었다. 눈금 막대는 50μM을 나타냅니다.
흥미롭게도, 10% FBS(pH =7.4)를 포함하는 DMEM 배지에서 hGC33-SFB-NP 및 SFB-NP 나노입자는 약 4일까지 빠른 약물 방출을 보인 다음, 상대적으로 느리고 안정적인 약물 방출을 보였습니다. 20일 동안 hGC33-SFB-NP 및 SFB-NP의 누적 SFB 방출은 각각 약 77% 및 65%였습니다(그림 1d). 차이는 PEG-b 표면의 친수성 분자 때문일 수 있습니다. - 수화를 증가시켜 가수분해를 촉진함으로써 나노입자의 분해를 촉진할 수 있는 PLGA 매트릭스. 제조된 NP의 안정성을 확인하기 위해 hGC33-SFB-NP, hGC33-null NP, null-NP 및 SFB-NP를 10% FBS(pH =7.4)가 포함된 DMEM 배지와 PBS(pH =7.4)에 넣었습니다. ). 다양한 NP의 크기는 2주 이상 안정적으로 유지되었습니다. SFB는 14일 동안 지속적이고 안정적인 방식으로 hGC33-SFB-NP에서 방출되었지만 10% FBS에서와 비교하여 DMEM 배지에서 입자 크기의 약간의 변화가 있었습니다(그림 1e, f). hGC33-SFB-NP의 안정성은 지속적인 생물학적 역할을 하는 SFB에 적합합니다.
큰 제타 전위는 NP 사이에 강한 정전기적 반발 상호작용을 일으키고 NP 분산 시스템의 안정성을 유지할 수 있다[23, 24]. 표 1에 나타난 바와 같이 hGC33-SFB-NP, hGC33-null-NP 및 SFB-NP의 제타 전위는 각각 − 18.2 ± 2.2mv, − 18.5 ± 1.8mv, .9mv입니다. 항체 hGC33 당단백질의 알데히드기, 카르복실기 및 인산기가 생성하는 음전하로 인해 발생합니다. 음전하를 띤 NP는 NP 현탁액의 높은 안정성으로 이어질 수 있습니다. 또한, 정상적인 생리적 환경에서는 세포 표면이 음전하를 띠기 때문에 제조된 나노입자는 낮은 전하로 세포를 밀어내고 조직과 세포에 대한 독성이 적다. 또한, null-NP와 SFB-NP(다분산도 지수[PDI] 0.18 및 0.19)의 크기 분포는 hGC33-null-NP(PDI =0.21) 및 hGC33-SFB-보다 약간 작지만 유의하게 작지는 않았습니다. NP(PDI =0.23). 따라서 나노입자의 크기는 균일성이 좋다. NP의 입자 크기, 입자 크기 분포 및 제타 전위는 SFB 캡슐화 효율 및 로딩 함량의 결과에 따라 표 1에 나타내었다. 초원심분리를 통해 접합되지 않은 GPC3 항체 hGC33을 제거하고, hGC33 항체의 NP에 대한 결합 효율을 평가하였다. BCA 단백질 분석 결과 hGC33 항체의 나노입자에 대한 결합 효율은 79.5% ± 2.9%였다.
hGC33-Coumarin 6-NP는 GPC3를 효과적으로 표적화합니다.
+HCC 세포주 HepG2 세포
나노입자의 hGC33 항체가 여전히 GPC3를 특이적으로 표적화하는 능력이 있는지 알아보기 위해 GPC3
+
를 사용했습니다. HepG2 및 GPC3
−
Li-7 세포를 표적 세포로, hGC33-coumarin 6-NP를 추적자 나노입자로 사용하고 서로 다른 세포를 2시간, 4시간, 8시간 동안 인큐베이션했습니다. 세포를 PBS로 3회 세척하고 DAPI와 반응시켜 핵을 염색하였다. 세포를 고정하고 Leica 형광현미경(DMi8, Germany)으로 촬영하였다. 동일한 배양 시간에 HepG2 세포의 녹색 형광이 Li-7 세포의 녹색 형광보다 유의하게 높은 것으로 나타났으며(그림 2), HepG2 세포에 들어가는 hGC33-coumarin 6-NP의 양이 그것보다 유의하게 높음을 나타냅니다. Li-7 세포에서. 결과는 hGC33-Coumarin6-NP에 대한 hGC33 항체가 여전히 GPC3을 표적으로 하고 나노입자의 내재화를 매개하는 능력이 있음을 문서화했습니다. HepG2 세포 및 Li-7 세포에서 GPC3의 발현을 간접 형광 및 세포화학적 염색으로 조사하였다. 그 결과 HepG2 세포는 GPC3를 과발현한 반면, Li-7 세포는 GPC3를 발현하지 않는 것으로 나타났다(그림 2).
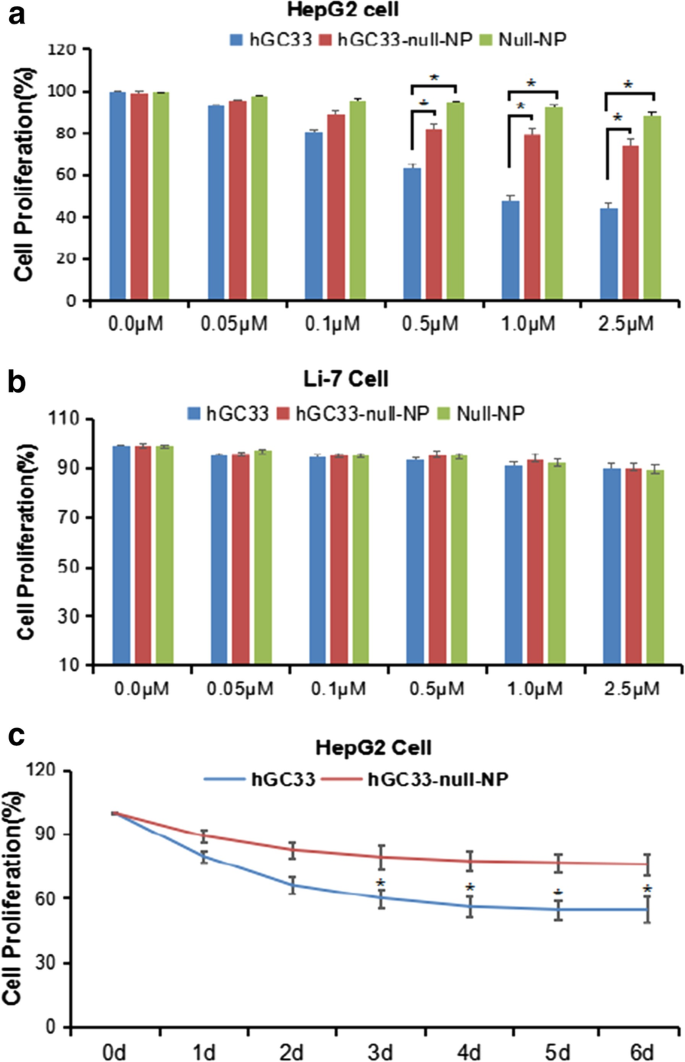
hGC33-Null-NP는 HepG2 세포의 증식을 억제합니다
hGC33으로 변형된 NP(hGC33-null-NP)가 간세포암종의 성장을 억제할 수 있는지 확인하기 위해 GPC3 양성 HCC 세포주 HepG2 및 GPC3 음성 세포주 Li-7의 세포 성장 억제를 측정했습니다. 우리는 hGC33-null-NP와 hGC33이 모두 24시간 처리 후 GPC3 양성 HCC 세포주 HepG2의 성장을 억제했지만 hGC33이 hGC33-null-NP보다 HepG2 세포에 대해 더 현저한 억제 효과가 있음을 발견했습니다(그림 3a).; 대조적으로, hGC33-null-NP 및 hGC33은 GPC3-음성 Li-7 세포주의 성장에 영향을 미치지 않았습니다(그림 3b). HepG2 세포 증식에 대한 hGC33-null-NP(0.1μm hgc33에 해당) 및 hGC33(0.1μm)의 대표적인 시간 경과는 그림 3c에 나와 있습니다. hGC33은 GPC3를 인식하는 C-말단 펩타이드이기 때문에 hGC33이 hGC33-null-NP보다 HepG2를 억제하는 데 우월하다. 결과는 hGC33-null-NP의 hGC33 분자가 공간 차단 효과를 가질 수 있으며, 이는 hGC33이 에피토프에 결합하는 능력에 영향을 미칠 수 있음을 시사합니다.
<사진>
HepG2 및 Li-7 세포의 증식은 hGC33, null-NP 및 hGC33-null-NP에 의해 억제되었다. 아 세포 증식 시험은 hGC33, null-NP 또는 hGC33-null-NP를 처리한 GPC3-양성 HepG2 세포에 대해 수행되었습니다. ㄴ 세포 증식 시험은 hGC33, null-NP 및 hGC33-null-NP를 처리한 GPC3 음성 Li-7 세포에 대해 수행되었습니다. 세포를 0~2.5μM hGC33, null-NP 또는 hGC33-null-NP와 함께 1일 동안 배양했습니다. 세포의 증식을 MTT 방법으로 측정하여 미처리 세포로 표준화하였다. 모든 값은 평균 ± SD를 나타냅니다. 항체 처리를 하지 않은 대조군(0μM)과 비교하여 *P <0.01; ㄷ hGC33 처리(1.0μM)에서 hGC33, null-NP 및 hGC33-null-NP에 대한 HepG2의 대표적인 반응 결과
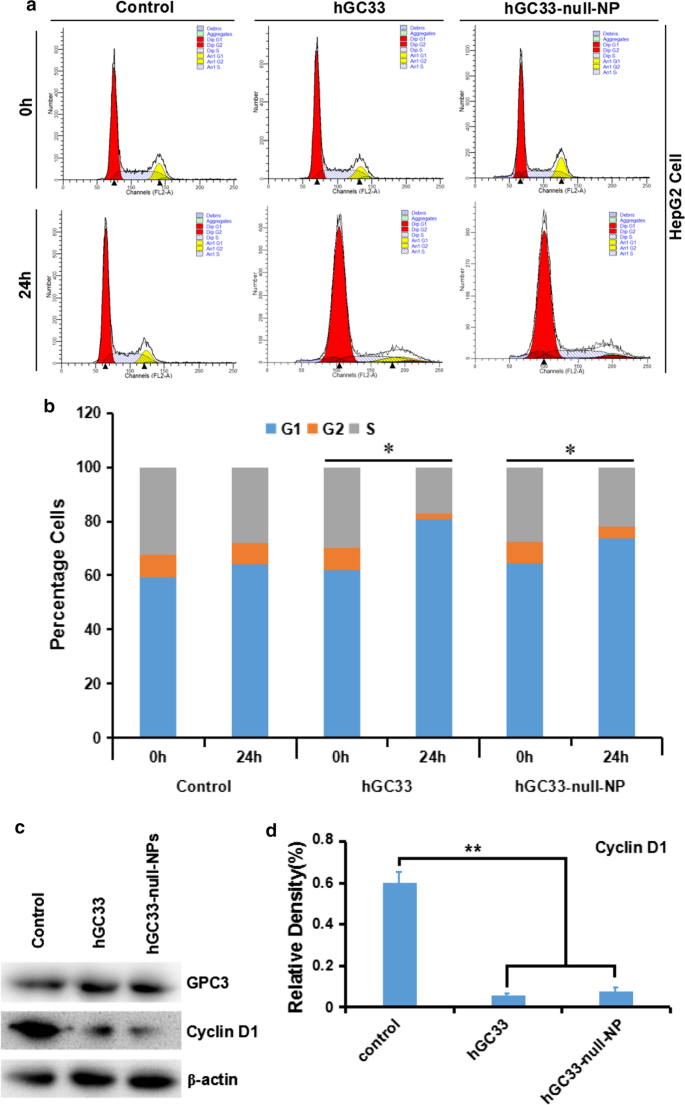
hGC33-Null-NP는 HepG2 세포의 세포 주기를 억제합니다.
hGC33-null-NP 나노입자에 변형된 hGC33 항체의 분자 활성 메커니즘을 이해하기 위해 hGC33-null-NP를 처리한 HepG2의 세포 주기 진행을 연구했습니다. HepG2 세포주에서 hGC33-null-NP와 hGC33은 G1에서 세포의 비율을 유의하게 증가시켰으며(그림 4a, b), 이는 hGC33-null-NP와 hGC33이 모두 G1 단계에서 세포 주기 정지를 유도할 수 있음을 나타냅니다. 또한, hGC33-null-NP 및 hGC33은 HepG2 세포에서 cyclinD1 발현을 유의하게 하향조절할 수 있었습니다(그림 4c, d).
<그림>
hGC33-null-NP 및 hGC33에 의해 유도된 세포 주기는 G1 단계에서 정지하고 HepG2 세포에서 cyclinD1 발현을 억제했습니다. 아 hGC33-null-NP 및 hGC33으로 처리된 다양한 그룹의 대표적인 세포 주기 다이어그램. ㄴ hGC33-null-NP 및 hGC33으로 처리된 다양한 그룹의 세포 주기 분석. HCC 세포를 0.5μm hGC33 또는 hGC33-null-NP(hGC33의 몰 농도를 기준으로 함)와 함께 배양했습니다. *피 <0.05, hGC33-null-NP 또는 hGC33 세포의 G1 단계를 0 h 세포의 G1 단계와 비교했습니다. ㄷ , d hGC33-null-NP 또는 hGC33 처리 후 cyclinD1은 대조군에 비해 HepG2 세포에서 유의하게 하향 조절되었습니다
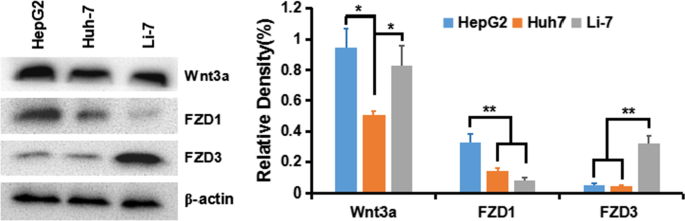
HepG2, Huh-7 및 Li-7 세포에서 Wnt 활성화
HCC 세포에서 고전적인 Wnt/β-Catenin 신호의 활성화를 이해하기 위해 우리는 먼저 다양한 HCC 세포주에서 Wnt 리간드 및 수용체 크림프 단백질(frizzled, FZD 또는 Frz)의 발현을 감지했습니다:HepG2(GPC3
++
), Huh-7(GPC
+
) 및 Li-7(GPC3
−
). 그 결과 β-Catenin 경로를 도입하는 Wnt3a 및 FZD 수용체가 모든 세포주, 특히 HepG2 및 Huh-7 세포에서 발현되는 것으로 나타났다(그림 5).
<그림>
HCC 세포주에서 Wnt3a, FZD1 및 FZD3의 발현
hGC33-Null-NP는 Wnt3a에 의해 유도된 Wnt 신호 전달 의존적 세포 증식을 억제합니다.
일부 연구에서는 GPC3의 세포외 부분이 Wnt/β-카테닌 신호 활성화 및 전달을 촉진하는 Wnt의 공동 수용체일 수 있음을 보여주었습니다. 따라서 HepG2(GPC3
+
), Huh-7(GPC3
+
) 및 Li-7(GPC3
−
) cells were co-incubated with hGC33 and hGC33-null-NP, the activation of Wnt/β-catenin signal was blocked by hGC33 and hGC33-null-NP, and proliferation of HepG2 and Huh-7, but not Li-7, in Wnt3a-conditioned medium was inhibited. That proliferation of Li-7 cells most likely is due to the absence of GPC3 on the surface of Li-7 cells (Fig. 6). Our results indicate hGC33 and hGC33-null-NP nanoparticles specifically bind to GPC3 molecules on cell membrane. hGC33 and hGC33-null-NP partially neutralize the mitogenic activity of Wnt3a and inhibit the Wnt/β-catenin signaling pathway. However, the inhibition of proliferation by hGC33-null-NP nanoparticles was less than that of hGC33. Perhaps the spatial structure of the nanoparticles interferes with hGC33-null-NP and limits the function of the hGC33 molecule on the nanoparticles so they cannot completely block the interaction between GPC3 and Wnt3a.
GPC3-activated Wnt signal transduction in HCC. Fifty percent Wnt3a DMEM medium was added with anti-wnt3a antibody, hGC33, or hGC33-null-NP. HepG2 (GPC3
++
), Huh-7 (GPC3
+
) and Li-7 (GPC3
−
) cells were co-incubated for 48 h, and cell proliferation was measured by MTT assay. The data were expressed as mean ± SD (*P < 0.01)
hGC33-Null-NP Inhibits Wnt3a-Induced Signal Transduction in HepG2 and Huh-7 Cells
To understand the effect of hGC33-null-NP on Wnt/β-catenin signaling in HCC cells, we extracted the proteins of HepG2 and Huh-7 cells treated with hGC33 or hGC33-null-NP. The results of western blot showed that after hGC33-null-NP treatment, the levels of pYAP and pβ-catenin were increased, but the levels of YAP and β-catenin were decreased. Furthermore, the levels of cyclinD1, CD44, VEGF, and c-MYC in the hGC33-null-NP group were lower than those in the control group, but the level was less than with hGC33 treatment. Similar effects were observed in HepG2 and Huh-7 cells, as shown in Fig. 7.
Inhibition of Wnt3a-induced β-catenin signaling by hGC33-null-NP or hGC33. 아 Compared with the control group, Wnt/β-catenin signaling pathway in HepG2 and Huh-7 cells treated with hGC33-null-NP or hGC33 was inhibited, and the levels of β-catenin and YAP were decreased, while the increased phosphorylated β-catenin and phosphorylated YAP molecules were unstable, and degraded in cytoplasm. The decreased β-catenin was difficult to maintain in the nucleus and drive the expression of CyclinD1, CD44, VEGF, and c-MYC, which resulted in the decrease of cyclinD1, CD44, VEGF, and c-myc protein levels. ㄴ The mechanism pattern of Wnt/β-catenin signaling pathway inhibited by hGC33-null-NP or hGC33
hGC33-SFB-NP or hGC33 Attenuates HCC Cell Migration by Inhibiting Epithelial Mesenchymal Transition (EMT)
HCC is one of the deadliest cancers in the world, and its incidence is steadily increasing. Sorafenib is the only approved standard treatment for patients with advanced HCC, as it has been shown to improve the survival rate of these patients. However, clinical and preclinical observations suggest that sorafenib therapy has limited efficacy due to the rapid development of drug resistance. Therefore, elucidating the mechanism of escape resistance to sorafenib is a major emphasis in HCC research. In recent years, more and more attention has been paid to the role of epithelial mesenchymal transition (EMT) in the progress of HCC and the development of drug resistance. EMT refers to the transformation of epithelial to mesenchymal cells, which endows cells with the ability metastasize and invade, including acquisition of stem cell characteristics, reducing apoptosis and aging, promoting immunosuppression, and participating in the occurrence and development of cancer. The loss of E-cadherin expression is considered a key step in the carcinogenesis and EMT. EMT is a developmental multi-step molecular and cellular reprogramming process that cancer cells use to achieve aggression. This is mainly through down regulating the expression of E-cadherin, keratin, mucin, ZO-1 (tight junction protein); up regulating the expression of vimentin, alpha-smooth muscle actin (α-SMA), FN fibronectin, MMPs (degradation matrix), N-cadherin, snail, slug, twist, Rho, TGF-β, FGF, type I collagen, and type II collagen to achieve the invasion, metastasis, and anti-apoptosis of EMT characteristic tumor. The changes of these protein expressions mainly involve the activation of Wnt/β-catenin and Ras/Raf/MAPK signaling pathways.
Our experiments have shown that hGC33 antibody on the surface of NP vector can inhibit Wnt3a-induced β-catenin signal transduction, and then down regulate the expression of β—catenin, CD44, vascular endothelial growth factor (VEGF), cyclin D1, and c-MYC. Furthermore, hGC33-SFB-NP inhibits the activation of Ras/Raf/MAPK signal pathway and inhibits proliferation and apoptosis of HCC cells. hGC33 and SFB have a synergistic effect, inhibiting EMT and decreasing the migration of HCC cells (Fig. 8).
Effect of hGC33-SFB-NP on EMT inhibition. 아 Compared with the control group, the hGC33-SFB-NP treatment group had less cell migration. Photographs were taken under an optical microscope (magnification, × 200). The error value represents the standard deviation of three independent experiments. *Compared with the control group, p < 0.01. ㄴ Compared with the control group, the EMT-related proteins snail, vimentin, and MMP-2 in HCCs treated with hGC33-SFB-NP decreased, whereas E-cadherin increased. ㄷ Molecular mechanism of EMT. EMT, epithelial–mesenchymal transition; MMP-2, matrix metalloproteinase-2; SFB, sorafenib; NP, nanoparticle
hGC33-SFB-NP Inhibits the Growth of Hepatocellular Carcinoma In Vivo and Improves the Survival Rate of Tumor-Bearing Mice
To evaluate the anti-tumor activity of hGC33-SFB-NP in vivo, HepG2 and Huh-7 cells were inoculated subcutaneously into the right abdomen and dorsal side of female BALB/c nude mice, respectively. When the tumor xenograft growth reached about 30 mm
3
, the mice were randomly divided into groups to further evaluate the inhibition of each group (hGC33-SFB-NP, hGC33-null-NP, SFB-NP, free hGC33, free SFB, and control group) HCC effect (n = 5 per group). It can be seen from Fig. 9a, b that hGC33-SFB-NP significantly slowed tumor growth in mice compared with the PBS control and other treatments. Compared with the PBS control, hGC33-null-NP, SFB-NP, free hGC33, and free SFB also had some inhibition of HCC, which is because free hGC33 and free SFB directly inhibit Wnt signal and Ras/Raf/MAPK, respectively. Such pathways can inhibit the proliferation of HCC cells to a certain extent. Although the nanoparticle-modified hGC33 (hGC33-null-NP) is connected to the nanosurface through chemical bonds, it did not affect hGC33′s targeting of GPC3 molecules and inhibition of Wnt activity. Nanoparticle-loaded SFB (SFB-NP), after being endocytosed by cells, was degraded to release SFB from the copolymer to inhibit the growth of HCC. In all, the inhibitory effect of hGC33-SFB-NP on HepG2 cell grafts was, as expected, more than on Huh-7 cell grafts, probably because HepG2 expresses GPC3 molecules.
The effect of hGC33-SFB-NP on xenotransplantation of HCC in nude mice and the changes of mice weight. Liver cancer cells were inoculated subcutaneously on the back of each nude mouse (n =10). After 10 days, the tumor bearing mice were treated with PBS (control), free hGC33, free SFB, hGC33-null-NP, SFB-NP, and hGC33-SFB-NP. Tumor size (a , b ) and body weight (c , d ) of mice were monitored at designated time points
The body weight of nude mice in each treatment group also was measured, as shown in Fig. 9c, d. The body weight of the control group decreased gradually. The weight of mice in free hGC33, free SFB, SFB-NP, and hGC33-null-NP treatment groups also decreased progressively and not significantly less than in the control group. However, the weight of nude mice bearing HepG2 and Huh-7 treated with hGC33-SFB-NP only slightly decreased, and the weight remained relatively stable during the treatment cycle. These results support that the novel hGC33-SFB-NP nanodrug has no significant toxicity in nude mice, and the SFB loaded on the nanocarrier and the surface modified hGC33 can produce additive or even synergistic anti-tumor effect.
Discussion
To examine the suitability of hGC33-SFB-NP for targeted HCC therapy, we tested the model conjugates for their ability to bind to human glypican-3 on HCC cells in vitro; to inhibit glypican-3-positive HCC cell proliferation, migration, and Wnt/β-catenin signal transduction; and inhibit HCC that overexpress glypican-3 in vivo.
To covalently bind GPC3-specific antibody hGC33 with mal-PEG-b -PLGA nanoparticles, we cross-linked the free sulfhydryl group in the Fc segment of hGC33 with maleimide functionalized PEG-b -PLGA (mal-PEG-b -PLGA) by forming stable thioether bonds. Conjugation is a prerequisite for targeting of GPC3-positive HCC. A series of experiments, including the changes of nanoparticle diameter and zeta potential detected by lens and the intracellular uptake of hGC33-SFB-NP, verified the targeting of hGC33-SFB-NP to HepG2 (GPC3
+
) 세포. These results indicated that the binding activity of antibody hGC33 was not altered by the conjugation.
We directly detected the phagocytic effect of GPC3
+
HepG2 and GPC3
−
Li-7 cells on PEG-b -PLGA NP surface-modified hGC33 by confocal microscopy. After HepG2 and Li-7 cells were co-cultured with hGC33-coumarin 6-NP, the green signal intensity in HepG2 cells was significantly higher than in Li-7 cells, indicating that there were more nanoparticles in the HepG2 cells. This finding is consistent with the hGC33 antibody modified on the surface of PEG-b -PLGA NP specifically binding to glypican-3 on the surface of HCC cells and being internalized. The efficiency of hGC33-modified NP internalization depends on the expression level of GPC3 antigen on the cell surface.
We used the standard MTT assay to measure the efficiency of inhibiting the proliferation of hepatoma cells. Both hGC33-null-NP and hGC33 inhibited the growth of the GPC3-positive HCC cell line HepG2, but hGC33-null-NP and hGC33 did not affect the proliferation of GPC3-negative Li-7 cells (Fig. 3b). At the animal level, hGC33-null-NP or hGC33 alone inhibited the growth of Huh-7 and HepG2 xenografts to a certain extent, while hGC33-SFB-NP caused growth arrest of Huh-7 and HepG2 hepatoma xenografts in mice. The finding that hGC33-null-NP significantly inhibited GPC3-positive hepatoma cells showed that the inhibitory effect of PEG-b -PLGA NP surface-modified hGC33 on HCC cell proliferation depends on the expression of GPC3 antigen on the cell surface.
GPC3 regulates many pathways in HCC pathogenesis, including Wnt and YAP signaling [25,26,27]. GPC3 interacts with Wnt ligand and may be a coreceptor for Wnt and facilitate Wnt/Frizzled binding for HCC growth [28, 29]. We further examined the effect of nanodrug surface-modified hGC33 on Wnt signaling pathway in hepatoma cells. Like free hGC33, nanodrug surface-modified hGC33 inhibited the proliferation of hepatoma cells not only by blocking Wnt-induced signal transduction in HepG2 and Huh-7 cells of expressing GPC3, but also by inhibiting Wnt3a-induced β-catenin and YAP signal transduction. Previous studies have shown that YAP expression is regulated by β-catenin at the transcriptional level of HCC [30, 31]. In this study, free hGC33 and nanodrug surface-modified hGC33 inhibited Wnt3a-induced YAP activity, indicating that Yap/TAZ released from β-catenin complex can also initiate classic Wnt signaling transduction [32, 2]. These results indicate that typical Wnt and YAP cross talk through a variety of mechanisms. Compared with hGC33-null-NP and hGC33, hGC33-SFB-NP had stronger anti-proliferation and anti-migration ability in vitro and in vivo. Thus, hGC33 not only determines the specificity of HCC cells, but also increases the inhibitory effect of SFB on the proliferation and migration of HCC cells by blocking the key signals related to tumor growth, such as Wnt/β-catenin and Wnt/YAP signaling pathway.
Conclusion
Antibody hGC33 to glypican-3, a membrane protein that is overexpressed on hepatocellular carcinoma cells, increased binding of sorafenib-loaded polyethylene glycol-b-PLGA polymer nanoparticles (hGC33-SFB-NP) to glypican-3 on the cancer cells. Administration of the antibody-modified nanoparticles synergistically inhibited Wnt-induced signal transduction and Ras/Raf/MAPK signaling pathway; hepatocellular carcinoma cells were arrested in G0/1 phase by down-regulation of cyclin D1 expression, thus attenuating cancer cell migration by inhibiting epithelial–mesenchymal transition. hGC33-SFB-NP inhibited the growth of liver cancer in vivo and improved the survival rate of tumor-bearing mice.